From Wikipedia, the free encyclopedia

Different types of indel mutation. Panel C is simply a deletion and not a frameshift mutation.
A frameshift mutation (also called a framing error or a reading frame shift) is a genetic mutation caused by indels (insertions or deletions) of a number of nucleotides in a DNA sequence that is not divisible by three. Due to the triplet nature of gene expression by codons, the insertion or deletion can change the reading frame (the grouping of the codons), resulting in a completely different translation from the original. The earlier in the sequence the deletion or insertion occurs, the more altered the protein.[1] A frameshift mutation is not the same as a single-nucleotide polymorphism in which a nucleotide is replaced, rather than inserted or deleted. A frameshift mutation will in general cause the reading of the codons after the mutation to code for different amino acids. The frameshift mutation will also alter the first stop codon («UAA», «UGA» or «UAG») encountered in the sequence. The polypeptide being created could be abnormally short or abnormally long, and will most likely not be functional.[2]
Frameshift mutations are apparent in severe genetic diseases such as Tay–Sachs disease; they increase susceptibility to certain cancers and classes of familial hypercholesterolaemia; in 1997,[3] a frameshift mutation was linked to resistance to infection by the HIV retrovirus. Frameshift mutations have been proposed as a source of biological novelty, as with the alleged creation of nylonase, however, this interpretation is controversial. A study by Negoro et al (2006)[4] found that a frameshift mutation was unlikely to have been the cause and that rather a two amino acid substitution in the active site of an ancestral esterase resulted in nylonase.
Background[edit]
The information contained in DNA determines protein function in the cells of all organisms. Transcription and translation allow this information to be communicated into making proteins. However, an error in reading this communication can cause protein function to be incorrect and eventually cause disease even as the cell incorporates a variety of corrective measures.
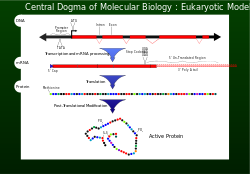
Central dogma[edit]
In 1956 Francis Crick described the flow of genetic information from DNA to a specific amino acid arrangement for making a protein as the central dogma.[1] For a cell to properly function, proteins are required to be produced accurately for structural and for catalytic activities. An incorrectly made protein can have detrimental effects on cell viability and in most cases cause the higher organism to become unhealthy by abnormal cellular functions. To ensure that the genome successfully passes the information on, proofreading mechanisms such as exonucleases and mismatch repair systems are incorporated in DNA replication .[1]
Transcription and translation[edit]
![]()
After DNA replication, the reading of a selected section of genetic information is accomplished by transcription.[1]
Nucleotides containing the genetic information are now on a single strand messenger template called mRNA. The mRNA is incorporated with a subunit of the ribosome and interacts with an rRNA. The genetic information carried in the codons of the mRNA are now read (decoded) by anticodons of the tRNA. As each codon (triplet) is read, amino acids are being joined together until a stop codon (UAG, UGA or UAA) is reached. At this point the polypeptide (protein) has been synthesised and is released.[1] For every 1000 amino acid incorporated into the protein, no more than one is incorrect. This fidelity of codon recognition, maintaining the importance of the proper reading frame, is accomplished by proper base pairing at the ribosome A site, GTP hydrolysis activity of EF-Tu a form of kinetic stability, and a proofreading mechanism as EF-Tu is released.[1]
Frameshifting may also occur during prophase translation, producing different proteins from overlapping open reading frames, such as the gag-pol-env retroviral proteins. This is fairly common in viruses and also occurs in bacteria and yeast (Farabaugh, 1996). Reverse transcriptase, as opposed to RNA Polymerase II, is thought to be a stronger cause of the occurrence of frameshift mutations. In experiments only 3–13% of all frameshift mutations occurred because of RNA Polymerase II. In prokaryotes the error rate inducing frameshift mutations is only somewhere in the range of .0001 and .00001.[5]
There are several biological processes that help to prevent frameshift mutations. Reverse mutations occur which change the mutated sequence back to the original wild type sequence. Another possibility for mutation correction is the use of a suppressor mutation. This offsets the effect of the original mutation by creating a secondary mutation, shifting the sequence to allow for the correct amino acids to be read. Guide RNA can also be used to insert or delete Uridine into the mRNA after transcription, this allows for the correct reading frame.[1]
Codon-triplet importance[edit]

The three letter code, the codon
A codon is a set of three nucleotides, a triplet that code for a certain amino acid. The first codon establishes the reading frame, whereby a new codon begins. A protein’s amino acid backbone sequence is defined by contiguous triplets.[6] Codons are key to translation of genetic information for the synthesis of proteins. The reading frame is set when translating the mRNA begins and is maintained as it reads one triplet to the next. The reading of the genetic code is subject to three rules the monitor codons in mRNA. First, codons are read in a 5′ to 3′ direction. Second, codons are nonoverlapping and the message has no gaps. The last rule, as stated above, that the message is translated in a fixed reading frame.[1]
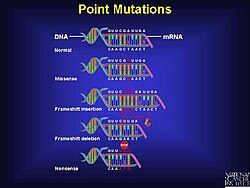
Example of different types of point mutations
Mechanism[edit]
Frameshift mutations can occur randomly or be caused by an external stimulus. The detection of frameshift mutations can occur via several different methods. Frameshifts are just one type of mutation that can lead to incomplete or incorrect proteins, but they account for a significant percentage of errors in DNA.
Genetic or environmental[edit]
This is a genetic mutation at the level of nucleotide bases. Why and how frameshift mutations occur are continually being sought after. An environmental study, specifically the production of UV-induced frameshift mutations by DNA polymerases deficient in 3′ → 5′ exonuclease activity was done. The normal sequence 5′ GTC GTT TTA CAA 3′ was changed to GTC GTT T TTA CAA (MIDT) of GTC GTT C TTA CAA (MIDC) to study frameshifts. E. coli pol I Kf and T7 DNA polymerase mutant enzymes devoid of 3′ → 5′ exonuclease activity produce UV-induced revertants at higher frequency than did their exonuclease proficient counterparts. The data indicates that loss of proofreading activity increases the frequency of UV-induced frameshifts.[7]
Detection[edit]
Fluorescence[edit]
The effects of neighboring bases and secondary structure to detect the frequency of frameshift mutations has been investigated in depth using fluorescence. Fluorescently tagged DNA, by means of base analogues, permits one to study the local changes of a DNA sequence.[8] Studies on the effects of the length of the primer strand reveal that an equilibrium mixture of four hybridization conformations was observed when template bases looped-out as a bulge, i.e. a structure flanked on both sides by duplex DNA. In contrast, a double-loop structure with an unusual unstacked DNA conformation at its downstream edge was observed when the extruded bases were positioned at the primer–template junction, showing that misalignments can be modified by neighboring DNA secondary structure.[9]
Sequencing[edit]
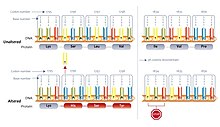
A deletion mutation alters every codon following it, and can make protein synthesis stop prematurely by forming a stop codon.
Sanger sequencing and pyrosequencing are two methods that have been used to detect frameshift mutations, however, it is likely that data generated will not be of the highest quality. Even still, 1.96 million indels have been identified through Sanger sequencing that do not overlap with other databases. When a frameshift mutation is observed it is compared against the Human Genome Mutation Database (HGMD) to determine if the mutation has a damaging effect. This is done by looking at four features. First, the ratio between the affected and conserved DNA, second the location of the mutation relative to the transcript, third the ratio of conserved and affected amino acids and finally the distance of the indel to the end of the exon.[10]
Massively Parallel Sequencing is a newer method that can be used to detect mutations. Using this method, up to 17 gigabases can be sequenced at once, as opposed to limited ranges for Sanger sequencing of only about 1 kilobase. Several technologies are available to perform this test and it is being looked at to be used in clinical applications.[11] When testing for different carcinomas, current methods only allow for looking at one gene at a time. Massively Parallel Sequencing can test for a variety of cancer causing mutations at once as opposed to several specific tests.[12] An experiment to determine the accuracy of this newer sequencing method tested for 21 genes and had no false positive calls for frameshift mutations.[13]
Diagnosis[edit]
A US patent (5,958,684) in 1999 by Leeuwen, details the methods and reagents for diagnosis of diseases caused by or associated with a gene having a somatic mutation giving rise to a frameshift mutation. The methods include providing a tissue or fluid sample and conducting gene analysis for frameshift mutation or a protein from this type of mutation. The nucleotide sequence of the suspected gene is provided from published gene sequences or from cloning and sequencing of the suspect gene. The amino acid sequence encoded by the gene is then predicted.[14]
Frequency[edit]
Despite the rules that govern the genetic code and the various mechanisms present in a cell to ensure the correct transfer of genetic information during the process of DNA replication as well as during translation, mutations do occur; frameshift mutation is not the only type. There are at least two other types of recognized point mutations, specifically missense mutation and nonsense mutation.[1] A frameshift mutation can drastically change the coding capacity (genetic information) of the message.[1] Small insertions or deletions (those less than 20 base pairs) make up 24% of mutations that manifest in currently recognized genetic disease.[10]
Frameshift mutations are found to be more common in repeat regions of DNA. A reason for this is because of slipping of the polymerase enzyme in repeat regions, allowing for mutations to enter the sequence.[15] Experiments can be run to determine the frequency of the frameshift mutation by adding or removing a pre-set number of nucleotides. Experiments have been run by adding four basepairs, called the +4 experiments, but a team from Emory University looked at the difference in frequency of the mutation by both adding and deleting a base pair. It was shown that there was no difference in the frequency between the addition and deletion of a base pair. There is however, a difference in the end result of the protein.[15]
Huntington’s disease is one of the nine codon reiteration disorders caused by polyglutamine expansion mutations that include spino-cerebellar ataxia (SCA) 1, 2, 6, 7 and 3, spinobulbar muscular atrophy and dentatorubal-pallidoluysianatrophy. There may be a link between diseases caused by polyglutamine and polyalanine expansion mutations, as frame shifting of the original SCA3 gene product encoding CAG/polyglutamines to GCA/polyalanines. Ribosomal slippage during translation of the SCA3 protein has been proposed as the mechanism resulting in shifting from the polyglutamine to the polyalanine-encoding frame. A dinucleotide deletion or single nucleotide insertion within the polyglutamine tract of huntingtin exon 1 would shift the CAG, polyglutamineen coding frame by +1 (+1 frame shift) to the GCA, polyalanine-encoding frame and introduce a novel epitope to the C terminus of Htt exon 1 (APAAAPAATRPGCG).[16]
Diseases[edit]
Several diseases have frameshift mutations as at least part of the cause. Knowing prevalent mutations can also aid in the diagnosis of the disease. Currently there are attempts to use frameshift mutations beneficially in the treatment of diseases, changing the reading frame of the amino acids.
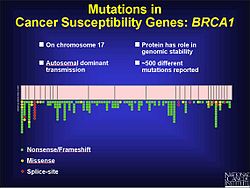
Frequency of mutations on BRCA1 gene on chromosome 17

Frequency of mutations on BRCA2 gene on chromosome 13
Cancer[edit]
Frameshift mutations are known to be a factor in colorectal cancer as well as other cancers with microsatellite instability. As stated previously, frameshift mutations are more likely to occur in a region of repeat sequence. When DNA mismatch repair does not fix the addition or deletion of bases, these mutations are more likely to be pathogenic. This may be in part because the tumor is not told to stop growing. Experiments in yeast and bacteria help to show characteristics of microsatellites that may contribute to defective DNA mismatch repair. These include the length of the microsatellite, the makeup of the genetic material and how pure the repeats are. Based on experimental results longer microsatellites have a higher rate of frameshift mutations. The flanking DNA can also contribute to frameshift mutations.[17] In prostate cancer a frameshift mutation changes the open reading frame (ORF) and prevents apoptosis from occurring. This leads to an unregulated growth of the tumor. While there are environmental factors that contribute to the progression of prostate cancer, there is also a genetic component. During testing of coding regions to identify mutations, 116 genetic variants were discovered, including 61 frameshift mutations.[18] There are over 500 mutations on chromosome 17 that seem to play a role in the development of breast and ovarian cancer in the BRCA1 gene, many of which are frameshift.[19]
Crohn’s disease[edit]
Crohn’s disease has an association with the NOD2 gene. The mutation is an insertion of a Cytosine at position 3020. This leads to a premature stop codon, shortening the protein that is supposed to be transcribed. When the protein is able to form normally, it responds to bacterial liposaccharides, where the 3020insC mutation prevents the protein from being responsive.[20]
Cystic fibrosis[edit]
Cystic fibrosis (CF) is a disease based on mutations in the CF transmembrane conductance regulator (CFTR) gene. There are over 1500 mutations identified, but not all cause the disease.[21] Most cases of cystic fibrosis are a result of the ∆F508 mutation, which deletes the entire amino acid. Two frameshift mutations are of interest in diagnosing CF, CF1213delT and CF1154-insTC. Both of these mutations commonly occur in tandem with at least one other mutation. They both lead to a small decrease in the function of the lungs and occur in about 1% of patients tested. These mutations were identified through Sanger sequencing.[22]
HIV[edit]
CCR5 is one of the cell entry co-factors associated with HIV, most frequently involved with nonsyncytium-inducing strains, is most apparent in HIV patients as opposed to AIDS patients. A 32 base pair deletion in CCR5 has been identified as a mutation that negates the likelihood of an HIV infection. This region on the open reading frame ORF contains a frameshift mutation leading to a premature stop codon. This leads to the loss of the HIV-coreceptor function in vitro. CCR5-1 is considered the wild type and CCR5-2 is considered to be the mutant allele. Those with a heterozygous mutation for the CCR5 were less susceptible to the development of HIV. In a study, despite high exposure to the HIV virus, there was no one homozygous for the CCR5 mutation that tested positive for HIV.[3]
Tay–Sachs disease[edit]
Tay–Sachs disease is a fatal disease affecting the central nervous system. It is most frequently found in infants and small children. Disease progression begins in the womb but symptoms do not appear until approximately 6 months of age. There is no cure for the disease.[23] Mutations in the β-hexosaminidase A (Hex A) gene are known to affect the onset of Tay-Sachs, with 78 mutations of different types being described, 67 of which are known to cause disease. Most of the mutations observed (65/78) are single base substitutions or SNPs, 11 deletions, 1 large and 10 small, and 2 insertions. 8 of the observed mutations are frameshift, 6 deletions and 2 insertions. A 4 base pair insertion in exon 11 is observed in 80% of Tay-Sachs disease presence in the Ashkenazi Jewish population. The frameshift mutations lead to an early stop codon which is known to play a role in the disease in infants. Delayed onset disease appears to be caused by 4 different mutations, one being a 3 base pair deletion.[24]
Smith–Magenis syndrome[edit]
Smith–Magenis syndrome (SMS) is a complex syndrome involving intellectual disabilities, sleep disturbance, behavioural problems, and a variety of craniofacial, skeletal, and visceral anomalies. The majority of SMS cases harbor an ~3.5 Mb common deletion that encompasses the retinoic acid induced-1 (RAI1) gene. Other cases illustrate variability in the SMS phenotype not previously shown for RAI1 mutation, including hearing loss, absence of self-abusive behaviours, and mild global delays. Sequencing of RAI1 revealed mutation of a heptamericC-tract (CCCCCCC) in exon 3 resulting in frameshift mutations. Of the seven reported frameshift mutations occurring in poly C-tracts in RAI1, four cases (~57%) occur at this heptameric C-tract. The results indicate that this heptameric C-tract is a preferential recombination hotspot insertion/deletions (SNindels) and therefore a primary target for analysis in patients suspected for mutations in RAI1.[25]
Hypertrophic cardiomyopathy[edit]
Hypertrophic cardiomyopathy is the most common cause of sudden death in young people, including trained athletes, and is caused by mutations in genes encoding proteins of the cardiac sarcomere. Mutations in the Troponin C gene (TNNC1) are a rare genetic cause of hypertrophic cardiomyopathy. A recent study has indicated that a frameshift mutation (c.363dupG or p.Gln122AlafsX30) in Troponin C was the cause of hypertrophic cardiomyopathy (and sudden cardiac death) in a 19-year-old male.[26]
Cures[edit]
Finding a cure for the diseases caused by frameshift mutations is rare. Research into this is ongoing. One example is a primary immunodeficiency (PID), an inherited condition which can lead to an increase in infections. There are 120 genes and 150 mutations that play a role in primary immunodeficiencies. The standard treatment is currently gene therapy, but this is a highly risky treatment and can often lead to other diseases, such as leukemia. Gene therapy procedures include modifying the zinc fringer nuclease fusion protein, cleaving both ends of the mutation, which in turn removes it from the sequence. Antisense-oligonucleotide mediated exon skipping is another possibility for Duchenne muscular dystrophy. This process allows for passing over the mutation so that the rest of the sequence remains in frame and the function of the protein stays intact. This, however, does not cure the disease, just treats symptoms, and is only practical in structural proteins or other repetitive genes. A third form of repair is revertant mosaicism, which is naturally occurring by creating a reverse mutation or a mutation at a second site that corrects the reading frame. This reversion may happen by intragenic recombination, mitotic gene conversion, second site DNA slipping or site-specific reversion. This is possible in several diseases, such as X-linked severe combined immunodeficiency (SCID), Wiskott–Aldrich syndrome, and Bloom syndrome. There are no drugs or other pharmacogenomic methods that help with PIDs.[27]
A European patent (EP1369126A1) in 2003 by Bork records a method used for prevention of cancers and for the curative treatment of cancers and precancers such as DNA-mismatch repair deficient (MMR) sporadic tumours and HNPCC associated tumours. The idea is to use immunotherapy with combinatorial mixtures of tumour-specific frameshift mutation-derived peptides to elicit a cytotoxic T-cell response specifically directed against tumour cells.[28]
See also[edit]
- Translational frameshift
- Mutation
- Transcription (genetics)
- Translation (biology)
- codon
- protein
- reading frame
- point mutation
- Crohn’s disease
- Tay–Sachs disease
References[edit]
- ^ a b c d e f g h i j Losick, Richard; Watson, James D.; Baker, Tania A.; Bell, Stephen; Gann, Alexander; Levine, Michael W. (2008). Molecular biology of the gene (6th ed.). San Francisco: Pearson/Benjamin Cummings. ISBN 978-0-8053-9592-1.
- ^ «DNA Is Constantly Changing through the Process of Mutation». Nature. Retrieved 17 May 2019.
- ^ a b Zimmerman PA, Buckler-White A, Alkhatib G, Spalding T, Kubofcik J, Combadiere C, Weissman D, Cohen O, Rubbert A, Lam G, Vaccarezza M, Kennedy PE, Kumaraswami V, Giorgi JV, Detels R, Hunter J, Chopek M, Berger EA, Fauci AS, Nutman TB, Murphy PM (January 1997). «Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk». Molecular Medicine (Cambridge, Mass.). 3 (1): 23–36. PMC 2230106. PMID 9132277.
- ^ Negoro S, Ohki T, Shibata N, Mizuno N, Wakitani Y, Tsurukame J, Matsumoto K, Kawamoto I, Takeo M, Higuchi Y (November 2005). «X-ray crystallographic analysis of 6-aminohexanoate-dimer hydrolase: molecular basis for the birth of a nylon oligomer-degrading enzyme». J Biol Chem. 280 (47): 39644–52. doi:10.1074/jbc.m505946200. PMID 16162506.
- ^ Zhang, J (August 2004). «Host RNA polymerase II makes minimal contributions to retroviral frame-shift mutations». The Journal of General Virology. 85 (Pt 8): 2389–95. doi:10.1099/vir.0.80081-0. PMID 15269381.
- ^ Cox, Michael; Nelson, David R.; Lehninger, Albert L (2008). Lehninger principles of biochemistry. San Francisco: W.H. Freeman. ISBN 978-0-7167-7108-1.
- ^ Sagher, Daphna; Turkington, Edith; Acharya, Sonia; Strauss, Bernard (July 1994). «Production of UV-induced Frameshift Mutations in Vitro by DNA Polymerases Deficient in 3′ → 5′ Exonuclease Activity». Journal of Molecular Biology. 240 (3): 226–242. doi:10.1006/jmbi.1994.1437. PMID 8028006.
- ^ Johnson, Neil P.; Walter A. Baase; Peter H. von Hippel (March 2004). «Low-energy circular dichroism of 2-aminopurine dinucleotide as a probe of local conformation of DNA and RNA». Proc Natl Acad Sci U S A. 101 (10): 3426–31. Bibcode:2004PNAS..101.3426J. doi:10.1073/pnas.0400591101. PMC 373478. PMID 14993592.
- ^ Baase, Walter A.; Davis Jose; Benjamin C. Ponedel; Peter H. von Hippel; Neil P. Johnson (2009). «DNA models of trinucleotide frameshift deletions: the formation of loops and bulges at the primer–template junction». Nucleic Acids Research. 37 (5): 1682–9. doi:10.1093/nar/gkn1042. PMC 2655659. PMID 19155277.
- ^ a b Hu, J; Ng, PC (9 February 2012). «Predicting the effects of frameshifting indels». Genome Biology. 13 (2): R9. doi:10.1186/gb-2012-13-2-r9. PMC 3334572. PMID 22322200.
- ^ Tucker, Tracy; Marra, Marco; Friedman, Jan M. (2009). «Massively Parallel Sequencing: The Next Big Thing in Genetic Medicine». The American Journal of Human Genetics. 85 (2): 142–154. doi:10.1016/j.ajhg.2009.06.022. PMC 2725244. PMID 19679224.
- ^ Walsh, T.; Casadei, S.; Lee, M. K.; Pennil, C. C.; Nord, A. S.; Thornton, A. M.; Roeb, W.; Agnew, K. J.; Stray, S. M.; Wickramanayake, A.; Norquist, B.; Pennington, K. P.; Garcia, R. L.; King, M.-C.; Swisher, E. M. (2011). «From the Cover: Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing». Proc Natl Acad Sci U S A. 108 (44): 18032–7. Bibcode:2011PNAS..10818032W. doi:10.1073/pnas.1115052108. PMC 3207658. PMID 22006311.
- ^ Walsh, T.; Lee, M. K.; Casadei, S.; Thornton, A. M.; Stray, S. M.; Pennil, C.; Nord, A. S.; Mandell, J. B.; Swisher, E. M.; King, M.-C. (2010). «Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing». Proc Natl Acad Sci U S A. 107 (28): 12629–33. Bibcode:2010PNAS..10712629W. doi:10.1073/pnas.1007983107. PMC 2906584. PMID 20616022.
- ^ US Patent 5,958,684 (September 28, 1999) «Diagnosis of Neurodegenerative Disease» by Leeuwen et al
- ^ a b Harfe, BD; Jinks-Robertson, S (July 1999). «Removal of frameshift intermediates by mismatch repair proteins in Saccharomyces cerevisiae». Molecular and Cellular Biology. 19 (7): 4766–73. doi:10.1128/MCB.19.7.4766. PMC 84275. PMID 10373526.
- ^ Davies, J E; Rubinsztein, D C (2006). «Polyalanine and polyserine frameshift products in Huntington’s disease». Journal of Medical Genetics. 43 (11): 893–896. doi:10.1136/jmg.2006.044222. PMC 2563184. PMID 16801344.
- ^ Schmoldt, A; Benthe, HF; Haberland, G (1 September 1975). «Digitoxin metabolism by rat liver microsomes». Biochemical Pharmacology. 24 (17): 1639–41. doi:10.1016/0006-2952(75)90094-5. PMID 10.
- ^ Xu, XiaoLin; Zhu, KaiChang; Liu, Feng; Wang, Yue; Shen, JianGuo; Jin, Jizhong; Wang, Zhong; Chen, Lin; Li, Jiadong; Xu, Min (May 2013). «Identification of somatic mutations in human prostate cancer by RNA-Seq». Gene. 519 (2): 343–7. doi:10.1016/j.gene.2013.01.046. PMID 23434521.
- ^ «Cancer Genomics». National Cancer Institute at the National Institute of Health. Archived from the original on 18 March 2013. Retrieved 24 March 2013.
- ^ Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nuñez G, Cho JH (May 31, 2001). «A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease» (PDF). Nature. 411 (6837): 603–6. Bibcode:2001Natur.411..603O. doi:10.1038/35079114. hdl:2027.42/62856. PMID 11385577. S2CID 205017657.
- ^ Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW (2008). «Guidelines for Diagnosis of Cystic Fibrosis in Newborns through Older Adults: Cystic Fibrosis Foundation Consensus Report». The Journal of Pediatrics. 153 (2): S4–S14. doi:10.1016/j.jpeds.2008.05.005. PMC 2810958. PMID 18639722.
- ^ Iannuzzi, MC; Stern, RC; Collins, FS; Hon, CT; Hidaka, N; Strong, T; Becker, L; Drumm, ML; White, MB; Gerrard, B (February 1991). «Two frameshift mutations in the cystic fibrosis gene». American Journal of Human Genetics. 48 (2): 227–31. PMC 1683026. PMID 1990834.
- ^ «Learning About Tay-Sachs Disease». National Human Genome Research Institute. Retrieved 24 March 2013.
- ^ Myerowitz, R (1997). «Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene». Human Mutation. 9 (3): 195–208. doi:10.1002/(SICI)1098-1004(1997)9:3<195::AID-HUMU1>3.0.CO;2-7. PMID 9090523.
- ^ Truong, Hoa T; Dudding, Tracy; Blanchard, Christopher L.; Elsea, Sarah H (2010). «Frameshift mutation hotspot identified in Smith-Magenis syndrome: case report and review of literature». BMC Medical Genetics. 11 (1): 142. doi:10.1186/1471-2350-11-142. PMC 2964533. PMID 20932317.
- ^ Chung WK, Kitner C, Maron BJ (June 2011). «Novel frameshift mutation in Troponin C ( TNNC1) associated with hypertrophic cardiomyopathy and sudden death». Cardiol Young. 21 (3): 345–8. doi:10.1017/S1047951110001927. PMID 21262074. S2CID 46682245.
- ^ Hu, Hailiang; Gatti, Richard A (2008). «New approaches to treatment of primary immunodeficiencies: fixing mutations with chemicals». Current Opinion in Allergy and Clinical Immunology. 8 (6): 540–6. doi:10.1097/ACI.0b013e328314b63b. PMC 2686128. PMID 18978469.
- ^ European Patent [1] (December 10, 2003) «Use of coding microsatellite region frameshift mutation-derived peptides for treating cancer» by Bork et al
Further reading[edit]
- Farabaugh PJ (1996). «Programmed translational frameshifting». Annu. Rev. Genet. 30 (1): 507–28. doi:10.1146/annurev.genet.30.1.507. PMC 239420. PMID 8982463.
- Lewis, Ricki (2005). Human Genetics: Concepts and Applications (6th ed.). Boston MA: McGraw Hill. pp. 227–8. ISBN 978-0-07-111156-0.
- «Nylonase Enzymes». 20 April 2004. Retrieved 2 June 2009.
External links[edit]
- Frameshift+Mutation at the US National Library of Medicine Medical Subject Headings (MeSH)
- NCBI dbSNP database — «a central repository for both single base nucleotide substitutions and short deletion and insertion polymorphisms»
- Wise2 — aligns a protein against a DNA sequence allowing frameshifts and introns
- FastY — compare a DNA sequence to a protein sequence database, allowing gaps and frameshifts
- Path — tool that compares two frameshift proteins (back-translation principle)
- HGMD — Human Genome Mutation Database
From Wikipedia, the free encyclopedia
Different types of indel mutation. Panel C is simply a deletion and not a frameshift mutation.
A frameshift mutation (also called a framing error or a reading frame shift) is a genetic mutation caused by indels (insertions or deletions) of a number of nucleotides in a DNA sequence that is not divisible by three. Due to the triplet nature of gene expression by codons, the insertion or deletion can change the reading frame (the grouping of the codons), resulting in a completely different translation from the original. The earlier in the sequence the deletion or insertion occurs, the more altered the protein.[1] A frameshift mutation is not the same as a single-nucleotide polymorphism in which a nucleotide is replaced, rather than inserted or deleted. A frameshift mutation will in general cause the reading of the codons after the mutation to code for different amino acids. The frameshift mutation will also alter the first stop codon («UAA», «UGA» or «UAG») encountered in the sequence. The polypeptide being created could be abnormally short or abnormally long, and will most likely not be functional.[2]
Frameshift mutations are apparent in severe genetic diseases such as Tay–Sachs disease; they increase susceptibility to certain cancers and classes of familial hypercholesterolaemia; in 1997,[3] a frameshift mutation was linked to resistance to infection by the HIV retrovirus. Frameshift mutations have been proposed as a source of biological novelty, as with the alleged creation of nylonase, however, this interpretation is controversial. A study by Negoro et al (2006)[4] found that a frameshift mutation was unlikely to have been the cause and that rather a two amino acid substitution in the active site of an ancestral esterase resulted in nylonase.
Background[edit]
The information contained in DNA determines protein function in the cells of all organisms. Transcription and translation allow this information to be communicated into making proteins. However, an error in reading this communication can cause protein function to be incorrect and eventually cause disease even as the cell incorporates a variety of corrective measures.
Central dogma[edit]
In 1956 Francis Crick described the flow of genetic information from DNA to a specific amino acid arrangement for making a protein as the central dogma.[1] For a cell to properly function, proteins are required to be produced accurately for structural and for catalytic activities. An incorrectly made protein can have detrimental effects on cell viability and in most cases cause the higher organism to become unhealthy by abnormal cellular functions. To ensure that the genome successfully passes the information on, proofreading mechanisms such as exonucleases and mismatch repair systems are incorporated in DNA replication .[1]
Transcription and translation[edit]
![]()
After DNA replication, the reading of a selected section of genetic information is accomplished by transcription.[1]
Nucleotides containing the genetic information are now on a single strand messenger template called mRNA. The mRNA is incorporated with a subunit of the ribosome and interacts with an rRNA. The genetic information carried in the codons of the mRNA are now read (decoded) by anticodons of the tRNA. As each codon (triplet) is read, amino acids are being joined together until a stop codon (UAG, UGA or UAA) is reached. At this point the polypeptide (protein) has been synthesised and is released.[1] For every 1000 amino acid incorporated into the protein, no more than one is incorrect. This fidelity of codon recognition, maintaining the importance of the proper reading frame, is accomplished by proper base pairing at the ribosome A site, GTP hydrolysis activity of EF-Tu a form of kinetic stability, and a proofreading mechanism as EF-Tu is released.[1]
Frameshifting may also occur during prophase translation, producing different proteins from overlapping open reading frames, such as the gag-pol-env retroviral proteins. This is fairly common in viruses and also occurs in bacteria and yeast (Farabaugh, 1996). Reverse transcriptase, as opposed to RNA Polymerase II, is thought to be a stronger cause of the occurrence of frameshift mutations. In experiments only 3–13% of all frameshift mutations occurred because of RNA Polymerase II. In prokaryotes the error rate inducing frameshift mutations is only somewhere in the range of .0001 and .00001.[5]
There are several biological processes that help to prevent frameshift mutations. Reverse mutations occur which change the mutated sequence back to the original wild type sequence. Another possibility for mutation correction is the use of a suppressor mutation. This offsets the effect of the original mutation by creating a secondary mutation, shifting the sequence to allow for the correct amino acids to be read. Guide RNA can also be used to insert or delete Uridine into the mRNA after transcription, this allows for the correct reading frame.[1]
Codon-triplet importance[edit]
The three letter code, the codon
A codon is a set of three nucleotides, a triplet that code for a certain amino acid. The first codon establishes the reading frame, whereby a new codon begins. A protein’s amino acid backbone sequence is defined by contiguous triplets.[6] Codons are key to translation of genetic information for the synthesis of proteins. The reading frame is set when translating the mRNA begins and is maintained as it reads one triplet to the next. The reading of the genetic code is subject to three rules the monitor codons in mRNA. First, codons are read in a 5′ to 3′ direction. Second, codons are nonoverlapping and the message has no gaps. The last rule, as stated above, that the message is translated in a fixed reading frame.[1]
Example of different types of point mutations
Mechanism[edit]
Frameshift mutations can occur randomly or be caused by an external stimulus. The detection of frameshift mutations can occur via several different methods. Frameshifts are just one type of mutation that can lead to incomplete or incorrect proteins, but they account for a significant percentage of errors in DNA.
Genetic or environmental[edit]
This is a genetic mutation at the level of nucleotide bases. Why and how frameshift mutations occur are continually being sought after. An environmental study, specifically the production of UV-induced frameshift mutations by DNA polymerases deficient in 3′ → 5′ exonuclease activity was done. The normal sequence 5′ GTC GTT TTA CAA 3′ was changed to GTC GTT T TTA CAA (MIDT) of GTC GTT C TTA CAA (MIDC) to study frameshifts. E. coli pol I Kf and T7 DNA polymerase mutant enzymes devoid of 3′ → 5′ exonuclease activity produce UV-induced revertants at higher frequency than did their exonuclease proficient counterparts. The data indicates that loss of proofreading activity increases the frequency of UV-induced frameshifts.[7]
Detection[edit]
Fluorescence[edit]
The effects of neighboring bases and secondary structure to detect the frequency of frameshift mutations has been investigated in depth using fluorescence. Fluorescently tagged DNA, by means of base analogues, permits one to study the local changes of a DNA sequence.[8] Studies on the effects of the length of the primer strand reveal that an equilibrium mixture of four hybridization conformations was observed when template bases looped-out as a bulge, i.e. a structure flanked on both sides by duplex DNA. In contrast, a double-loop structure with an unusual unstacked DNA conformation at its downstream edge was observed when the extruded bases were positioned at the primer–template junction, showing that misalignments can be modified by neighboring DNA secondary structure.[9]
Sequencing[edit]
A deletion mutation alters every codon following it, and can make protein synthesis stop prematurely by forming a stop codon.
Sanger sequencing and pyrosequencing are two methods that have been used to detect frameshift mutations, however, it is likely that data generated will not be of the highest quality. Even still, 1.96 million indels have been identified through Sanger sequencing that do not overlap with other databases. When a frameshift mutation is observed it is compared against the Human Genome Mutation Database (HGMD) to determine if the mutation has a damaging effect. This is done by looking at four features. First, the ratio between the affected and conserved DNA, second the location of the mutation relative to the transcript, third the ratio of conserved and affected amino acids and finally the distance of the indel to the end of the exon.[10]
Massively Parallel Sequencing is a newer method that can be used to detect mutations. Using this method, up to 17 gigabases can be sequenced at once, as opposed to limited ranges for Sanger sequencing of only about 1 kilobase. Several technologies are available to perform this test and it is being looked at to be used in clinical applications.[11] When testing for different carcinomas, current methods only allow for looking at one gene at a time. Massively Parallel Sequencing can test for a variety of cancer causing mutations at once as opposed to several specific tests.[12] An experiment to determine the accuracy of this newer sequencing method tested for 21 genes and had no false positive calls for frameshift mutations.[13]
Diagnosis[edit]
A US patent (5,958,684) in 1999 by Leeuwen, details the methods and reagents for diagnosis of diseases caused by or associated with a gene having a somatic mutation giving rise to a frameshift mutation. The methods include providing a tissue or fluid sample and conducting gene analysis for frameshift mutation or a protein from this type of mutation. The nucleotide sequence of the suspected gene is provided from published gene sequences or from cloning and sequencing of the suspect gene. The amino acid sequence encoded by the gene is then predicted.[14]
Frequency[edit]
Despite the rules that govern the genetic code and the various mechanisms present in a cell to ensure the correct transfer of genetic information during the process of DNA replication as well as during translation, mutations do occur; frameshift mutation is not the only type. There are at least two other types of recognized point mutations, specifically missense mutation and nonsense mutation.[1] A frameshift mutation can drastically change the coding capacity (genetic information) of the message.[1] Small insertions or deletions (those less than 20 base pairs) make up 24% of mutations that manifest in currently recognized genetic disease.[10]
Frameshift mutations are found to be more common in repeat regions of DNA. A reason for this is because of slipping of the polymerase enzyme in repeat regions, allowing for mutations to enter the sequence.[15] Experiments can be run to determine the frequency of the frameshift mutation by adding or removing a pre-set number of nucleotides. Experiments have been run by adding four basepairs, called the +4 experiments, but a team from Emory University looked at the difference in frequency of the mutation by both adding and deleting a base pair. It was shown that there was no difference in the frequency between the addition and deletion of a base pair. There is however, a difference in the end result of the protein.[15]
Huntington’s disease is one of the nine codon reiteration disorders caused by polyglutamine expansion mutations that include spino-cerebellar ataxia (SCA) 1, 2, 6, 7 and 3, spinobulbar muscular atrophy and dentatorubal-pallidoluysianatrophy. There may be a link between diseases caused by polyglutamine and polyalanine expansion mutations, as frame shifting of the original SCA3 gene product encoding CAG/polyglutamines to GCA/polyalanines. Ribosomal slippage during translation of the SCA3 protein has been proposed as the mechanism resulting in shifting from the polyglutamine to the polyalanine-encoding frame. A dinucleotide deletion or single nucleotide insertion within the polyglutamine tract of huntingtin exon 1 would shift the CAG, polyglutamineen coding frame by +1 (+1 frame shift) to the GCA, polyalanine-encoding frame and introduce a novel epitope to the C terminus of Htt exon 1 (APAAAPAATRPGCG).[16]
Diseases[edit]
Several diseases have frameshift mutations as at least part of the cause. Knowing prevalent mutations can also aid in the diagnosis of the disease. Currently there are attempts to use frameshift mutations beneficially in the treatment of diseases, changing the reading frame of the amino acids.
Frequency of mutations on BRCA1 gene on chromosome 17
Frequency of mutations on BRCA2 gene on chromosome 13
Cancer[edit]
Frameshift mutations are known to be a factor in colorectal cancer as well as other cancers with microsatellite instability. As stated previously, frameshift mutations are more likely to occur in a region of repeat sequence. When DNA mismatch repair does not fix the addition or deletion of bases, these mutations are more likely to be pathogenic. This may be in part because the tumor is not told to stop growing. Experiments in yeast and bacteria help to show characteristics of microsatellites that may contribute to defective DNA mismatch repair. These include the length of the microsatellite, the makeup of the genetic material and how pure the repeats are. Based on experimental results longer microsatellites have a higher rate of frameshift mutations. The flanking DNA can also contribute to frameshift mutations.[17] In prostate cancer a frameshift mutation changes the open reading frame (ORF) and prevents apoptosis from occurring. This leads to an unregulated growth of the tumor. While there are environmental factors that contribute to the progression of prostate cancer, there is also a genetic component. During testing of coding regions to identify mutations, 116 genetic variants were discovered, including 61 frameshift mutations.[18] There are over 500 mutations on chromosome 17 that seem to play a role in the development of breast and ovarian cancer in the BRCA1 gene, many of which are frameshift.[19]
Crohn’s disease[edit]
Crohn’s disease has an association with the NOD2 gene. The mutation is an insertion of a Cytosine at position 3020. This leads to a premature stop codon, shortening the protein that is supposed to be transcribed. When the protein is able to form normally, it responds to bacterial liposaccharides, where the 3020insC mutation prevents the protein from being responsive.[20]
Cystic fibrosis[edit]
Cystic fibrosis (CF) is a disease based on mutations in the CF transmembrane conductance regulator (CFTR) gene. There are over 1500 mutations identified, but not all cause the disease.[21] Most cases of cystic fibrosis are a result of the ∆F508 mutation, which deletes the entire amino acid. Two frameshift mutations are of interest in diagnosing CF, CF1213delT and CF1154-insTC. Both of these mutations commonly occur in tandem with at least one other mutation. They both lead to a small decrease in the function of the lungs and occur in about 1% of patients tested. These mutations were identified through Sanger sequencing.[22]
HIV[edit]
CCR5 is one of the cell entry co-factors associated with HIV, most frequently involved with nonsyncytium-inducing strains, is most apparent in HIV patients as opposed to AIDS patients. A 32 base pair deletion in CCR5 has been identified as a mutation that negates the likelihood of an HIV infection. This region on the open reading frame ORF contains a frameshift mutation leading to a premature stop codon. This leads to the loss of the HIV-coreceptor function in vitro. CCR5-1 is considered the wild type and CCR5-2 is considered to be the mutant allele. Those with a heterozygous mutation for the CCR5 were less susceptible to the development of HIV. In a study, despite high exposure to the HIV virus, there was no one homozygous for the CCR5 mutation that tested positive for HIV.[3]
Tay–Sachs disease[edit]
Tay–Sachs disease is a fatal disease affecting the central nervous system. It is most frequently found in infants and small children. Disease progression begins in the womb but symptoms do not appear until approximately 6 months of age. There is no cure for the disease.[23] Mutations in the β-hexosaminidase A (Hex A) gene are known to affect the onset of Tay-Sachs, with 78 mutations of different types being described, 67 of which are known to cause disease. Most of the mutations observed (65/78) are single base substitutions or SNPs, 11 deletions, 1 large and 10 small, and 2 insertions. 8 of the observed mutations are frameshift, 6 deletions and 2 insertions. A 4 base pair insertion in exon 11 is observed in 80% of Tay-Sachs disease presence in the Ashkenazi Jewish population. The frameshift mutations lead to an early stop codon which is known to play a role in the disease in infants. Delayed onset disease appears to be caused by 4 different mutations, one being a 3 base pair deletion.[24]
Smith–Magenis syndrome[edit]
Smith–Magenis syndrome (SMS) is a complex syndrome involving intellectual disabilities, sleep disturbance, behavioural problems, and a variety of craniofacial, skeletal, and visceral anomalies. The majority of SMS cases harbor an ~3.5 Mb common deletion that encompasses the retinoic acid induced-1 (RAI1) gene. Other cases illustrate variability in the SMS phenotype not previously shown for RAI1 mutation, including hearing loss, absence of self-abusive behaviours, and mild global delays. Sequencing of RAI1 revealed mutation of a heptamericC-tract (CCCCCCC) in exon 3 resulting in frameshift mutations. Of the seven reported frameshift mutations occurring in poly C-tracts in RAI1, four cases (~57%) occur at this heptameric C-tract. The results indicate that this heptameric C-tract is a preferential recombination hotspot insertion/deletions (SNindels) and therefore a primary target for analysis in patients suspected for mutations in RAI1.[25]
Hypertrophic cardiomyopathy[edit]
Hypertrophic cardiomyopathy is the most common cause of sudden death in young people, including trained athletes, and is caused by mutations in genes encoding proteins of the cardiac sarcomere. Mutations in the Troponin C gene (TNNC1) are a rare genetic cause of hypertrophic cardiomyopathy. A recent study has indicated that a frameshift mutation (c.363dupG or p.Gln122AlafsX30) in Troponin C was the cause of hypertrophic cardiomyopathy (and sudden cardiac death) in a 19-year-old male.[26]
Cures[edit]
Finding a cure for the diseases caused by frameshift mutations is rare. Research into this is ongoing. One example is a primary immunodeficiency (PID), an inherited condition which can lead to an increase in infections. There are 120 genes and 150 mutations that play a role in primary immunodeficiencies. The standard treatment is currently gene therapy, but this is a highly risky treatment and can often lead to other diseases, such as leukemia. Gene therapy procedures include modifying the zinc fringer nuclease fusion protein, cleaving both ends of the mutation, which in turn removes it from the sequence. Antisense-oligonucleotide mediated exon skipping is another possibility for Duchenne muscular dystrophy. This process allows for passing over the mutation so that the rest of the sequence remains in frame and the function of the protein stays intact. This, however, does not cure the disease, just treats symptoms, and is only practical in structural proteins or other repetitive genes. A third form of repair is revertant mosaicism, which is naturally occurring by creating a reverse mutation or a mutation at a second site that corrects the reading frame. This reversion may happen by intragenic recombination, mitotic gene conversion, second site DNA slipping or site-specific reversion. This is possible in several diseases, such as X-linked severe combined immunodeficiency (SCID), Wiskott–Aldrich syndrome, and Bloom syndrome. There are no drugs or other pharmacogenomic methods that help with PIDs.[27]
A European patent (EP1369126A1) in 2003 by Bork records a method used for prevention of cancers and for the curative treatment of cancers and precancers such as DNA-mismatch repair deficient (MMR) sporadic tumours and HNPCC associated tumours. The idea is to use immunotherapy with combinatorial mixtures of tumour-specific frameshift mutation-derived peptides to elicit a cytotoxic T-cell response specifically directed against tumour cells.[28]
See also[edit]
- Translational frameshift
- Mutation
- Transcription (genetics)
- Translation (biology)
- codon
- protein
- reading frame
- point mutation
- Crohn’s disease
- Tay–Sachs disease
References[edit]
- ^ a b c d e f g h i j Losick, Richard; Watson, James D.; Baker, Tania A.; Bell, Stephen; Gann, Alexander; Levine, Michael W. (2008). Molecular biology of the gene (6th ed.). San Francisco: Pearson/Benjamin Cummings. ISBN 978-0-8053-9592-1.
- ^ «DNA Is Constantly Changing through the Process of Mutation». Nature. Retrieved 17 May 2019.
- ^ a b Zimmerman PA, Buckler-White A, Alkhatib G, Spalding T, Kubofcik J, Combadiere C, Weissman D, Cohen O, Rubbert A, Lam G, Vaccarezza M, Kennedy PE, Kumaraswami V, Giorgi JV, Detels R, Hunter J, Chopek M, Berger EA, Fauci AS, Nutman TB, Murphy PM (January 1997). «Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk». Molecular Medicine (Cambridge, Mass.). 3 (1): 23–36. PMC 2230106. PMID 9132277.
- ^ Negoro S, Ohki T, Shibata N, Mizuno N, Wakitani Y, Tsurukame J, Matsumoto K, Kawamoto I, Takeo M, Higuchi Y (November 2005). «X-ray crystallographic analysis of 6-aminohexanoate-dimer hydrolase: molecular basis for the birth of a nylon oligomer-degrading enzyme». J Biol Chem. 280 (47): 39644–52. doi:10.1074/jbc.m505946200. PMID 16162506.
- ^ Zhang, J (August 2004). «Host RNA polymerase II makes minimal contributions to retroviral frame-shift mutations». The Journal of General Virology. 85 (Pt 8): 2389–95. doi:10.1099/vir.0.80081-0. PMID 15269381.
- ^ Cox, Michael; Nelson, David R.; Lehninger, Albert L (2008). Lehninger principles of biochemistry. San Francisco: W.H. Freeman. ISBN 978-0-7167-7108-1.
- ^ Sagher, Daphna; Turkington, Edith; Acharya, Sonia; Strauss, Bernard (July 1994). «Production of UV-induced Frameshift Mutations in Vitro by DNA Polymerases Deficient in 3′ → 5′ Exonuclease Activity». Journal of Molecular Biology. 240 (3): 226–242. doi:10.1006/jmbi.1994.1437. PMID 8028006.
- ^ Johnson, Neil P.; Walter A. Baase; Peter H. von Hippel (March 2004). «Low-energy circular dichroism of 2-aminopurine dinucleotide as a probe of local conformation of DNA and RNA». Proc Natl Acad Sci U S A. 101 (10): 3426–31. Bibcode:2004PNAS..101.3426J. doi:10.1073/pnas.0400591101. PMC 373478. PMID 14993592.
- ^ Baase, Walter A.; Davis Jose; Benjamin C. Ponedel; Peter H. von Hippel; Neil P. Johnson (2009). «DNA models of trinucleotide frameshift deletions: the formation of loops and bulges at the primer–template junction». Nucleic Acids Research. 37 (5): 1682–9. doi:10.1093/nar/gkn1042. PMC 2655659. PMID 19155277.
- ^ a b Hu, J; Ng, PC (9 February 2012). «Predicting the effects of frameshifting indels». Genome Biology. 13 (2): R9. doi:10.1186/gb-2012-13-2-r9. PMC 3334572. PMID 22322200.
- ^ Tucker, Tracy; Marra, Marco; Friedman, Jan M. (2009). «Massively Parallel Sequencing: The Next Big Thing in Genetic Medicine». The American Journal of Human Genetics. 85 (2): 142–154. doi:10.1016/j.ajhg.2009.06.022. PMC 2725244. PMID 19679224.
- ^ Walsh, T.; Casadei, S.; Lee, M. K.; Pennil, C. C.; Nord, A. S.; Thornton, A. M.; Roeb, W.; Agnew, K. J.; Stray, S. M.; Wickramanayake, A.; Norquist, B.; Pennington, K. P.; Garcia, R. L.; King, M.-C.; Swisher, E. M. (2011). «From the Cover: Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing». Proc Natl Acad Sci U S A. 108 (44): 18032–7. Bibcode:2011PNAS..10818032W. doi:10.1073/pnas.1115052108. PMC 3207658. PMID 22006311.
- ^ Walsh, T.; Lee, M. K.; Casadei, S.; Thornton, A. M.; Stray, S. M.; Pennil, C.; Nord, A. S.; Mandell, J. B.; Swisher, E. M.; King, M.-C. (2010). «Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing». Proc Natl Acad Sci U S A. 107 (28): 12629–33. Bibcode:2010PNAS..10712629W. doi:10.1073/pnas.1007983107. PMC 2906584. PMID 20616022.
- ^ US Patent 5,958,684 (September 28, 1999) «Diagnosis of Neurodegenerative Disease» by Leeuwen et al
- ^ a b Harfe, BD; Jinks-Robertson, S (July 1999). «Removal of frameshift intermediates by mismatch repair proteins in Saccharomyces cerevisiae». Molecular and Cellular Biology. 19 (7): 4766–73. doi:10.1128/MCB.19.7.4766. PMC 84275. PMID 10373526.
- ^ Davies, J E; Rubinsztein, D C (2006). «Polyalanine and polyserine frameshift products in Huntington’s disease». Journal of Medical Genetics. 43 (11): 893–896. doi:10.1136/jmg.2006.044222. PMC 2563184. PMID 16801344.
- ^ Schmoldt, A; Benthe, HF; Haberland, G (1 September 1975). «Digitoxin metabolism by rat liver microsomes». Biochemical Pharmacology. 24 (17): 1639–41. doi:10.1016/0006-2952(75)90094-5. PMID 10.
- ^ Xu, XiaoLin; Zhu, KaiChang; Liu, Feng; Wang, Yue; Shen, JianGuo; Jin, Jizhong; Wang, Zhong; Chen, Lin; Li, Jiadong; Xu, Min (May 2013). «Identification of somatic mutations in human prostate cancer by RNA-Seq». Gene. 519 (2): 343–7. doi:10.1016/j.gene.2013.01.046. PMID 23434521.
- ^ «Cancer Genomics». National Cancer Institute at the National Institute of Health. Archived from the original on 18 March 2013. Retrieved 24 March 2013.
- ^ Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nuñez G, Cho JH (May 31, 2001). «A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease» (PDF). Nature. 411 (6837): 603–6. Bibcode:2001Natur.411..603O. doi:10.1038/35079114. hdl:2027.42/62856. PMID 11385577. S2CID 205017657.
- ^ Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW (2008). «Guidelines for Diagnosis of Cystic Fibrosis in Newborns through Older Adults: Cystic Fibrosis Foundation Consensus Report». The Journal of Pediatrics. 153 (2): S4–S14. doi:10.1016/j.jpeds.2008.05.005. PMC 2810958. PMID 18639722.
- ^ Iannuzzi, MC; Stern, RC; Collins, FS; Hon, CT; Hidaka, N; Strong, T; Becker, L; Drumm, ML; White, MB; Gerrard, B (February 1991). «Two frameshift mutations in the cystic fibrosis gene». American Journal of Human Genetics. 48 (2): 227–31. PMC 1683026. PMID 1990834.
- ^ «Learning About Tay-Sachs Disease». National Human Genome Research Institute. Retrieved 24 March 2013.
- ^ Myerowitz, R (1997). «Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene». Human Mutation. 9 (3): 195–208. doi:10.1002/(SICI)1098-1004(1997)9:3<195::AID-HUMU1>3.0.CO;2-7. PMID 9090523.
- ^ Truong, Hoa T; Dudding, Tracy; Blanchard, Christopher L.; Elsea, Sarah H (2010). «Frameshift mutation hotspot identified in Smith-Magenis syndrome: case report and review of literature». BMC Medical Genetics. 11 (1): 142. doi:10.1186/1471-2350-11-142. PMC 2964533. PMID 20932317.
- ^ Chung WK, Kitner C, Maron BJ (June 2011). «Novel frameshift mutation in Troponin C ( TNNC1) associated with hypertrophic cardiomyopathy and sudden death». Cardiol Young. 21 (3): 345–8. doi:10.1017/S1047951110001927. PMID 21262074. S2CID 46682245.
- ^ Hu, Hailiang; Gatti, Richard A (2008). «New approaches to treatment of primary immunodeficiencies: fixing mutations with chemicals». Current Opinion in Allergy and Clinical Immunology. 8 (6): 540–6. doi:10.1097/ACI.0b013e328314b63b. PMC 2686128. PMID 18978469.
- ^ European Patent [1] (December 10, 2003) «Use of coding microsatellite region frameshift mutation-derived peptides for treating cancer» by Bork et al
Further reading[edit]
- Farabaugh PJ (1996). «Programmed translational frameshifting». Annu. Rev. Genet. 30 (1): 507–28. doi:10.1146/annurev.genet.30.1.507. PMC 239420. PMID 8982463.
- Lewis, Ricki (2005). Human Genetics: Concepts and Applications (6th ed.). Boston MA: McGraw Hill. pp. 227–8. ISBN 978-0-07-111156-0.
- «Nylonase Enzymes». 20 April 2004. Retrieved 2 June 2009.
External links[edit]
- Frameshift+Mutation at the US National Library of Medicine Medical Subject Headings (MeSH)
- NCBI dbSNP database — «a central repository for both single base nucleotide substitutions and short deletion and insertion polymorphisms»
- Wise2 — aligns a protein against a DNA sequence allowing frameshifts and introns
- FastY — compare a DNA sequence to a protein sequence database, allowing gaps and frameshifts
- Path — tool that compares two frameshift proteins (back-translation principle)
- HGMD — Human Genome Mutation Database
Молекулярная биология клетки — Том 1 — Албертс Б., Брей Д., Льюис Дж., Рэфф М., Робертс К., Уотсон Дж. 1994
Молекулярная организация клеток
Основные генетические механизмы
Механизмы репликации ДНК
Живые организмы должны не только поддерживать целостность нуклеотидных последовательностей ДНК путем ее репарации, но еще и очень точно воспроизводить свою ДНК перед каждым клеточным делением. При репликации ДНК скорость полимеризации колеблется в пределах от 500 нуклеотидов в 1 с у бактерий приблизительно до 50 нуклеотидов у млекопитающих. Ясно, что ферменты, катализирующие процесс репликации, должны работать и точно, и быстро. Быстрота и точность достигаются с помощью особого мультиферментного комплекса, направляющего процесс репликации. Этот комплекс, состоящий из нескольких различных белков, представляет собой сложный и совершенный «аппарат репликации».
5.3.1. Репликация ДНК, как и ее репарация, основана на комплементарном спаривании оснований [25]
Матричная активность ДНК проявляется в том, что ее нуклеотидная последовательность копируется (целиком или частично) путем комплементарного спаривания оснований (А с Т или G с С) в виде комплементарной последовательности нуклеотидов ДНК или РНК. Этот процесс предполагает узнавание каждого нуклеотида в ДНК свободным (неполимеризованным) комплементарным нуклеотидом и обязательное разделение (хотя бы на время) двух цепей ДНК, с тем чтобы в каждом основании группы, играющие роль доноров и акцепторов при образовании водородных связей, оказались доступными для комплементарного спаривания. Таким образом поступающие одиночные нуклеотиды выстраиваются в определенном порядке вдоль матричной цепи ДНК для ферментативной полимеризации, продуктом которой является новая полинуклеотидная цепь. В 1957 г. был открыт первый фермент, катализирующий процесс полимеризации нуклеотидов; он был назван ДНК-полимеразой. Было показано, что субстратами ДНК-полимеразы служат дезоксирибонуклеозидтрифосфаты, полимеризующиеся на одно-цепочечной ДНК-матрице (двухступенчатый механизм этой полимеризации представлен на рис. 5-34 в связи с обсуждением процесса репарации ДНК). Позже была выделена и РНК-полимераза, для которой субстратами служат рибонуклеозидтрифосфаты.
Во время репликации ДНК каждая из двух ее старых цепей служит матрицей для образования новой цепи. Поэтому чрезвычайно длинная нуклеотидная последовательность клеточной ДНК реплицируется, как это принято называть, «полуконсервативно» и каждая из двух дочерних клеток получает при клеточном делении новую двойную спираль ДНК, состоящую из одной старой и одной новой цепи (см. рис. 3-11).
5-23
5.3.2. Репликационная вилка асимметрична [26]
Исследования, проведенные в начале 1960-х годов на реплицирующихся хромосомах, в которые в качестве импульсной метки вводили радиоактивный предшественник ДНК 3Н-тимидин, выявили особую четко ограниченную область репликации, перемещающуюся вдоль родительской спирали ДНК. Эта активная область из-за своей Y-образной формы была названа репликационной вилкой. Именно в ней с помощью мультиферментного комплекса, содержащего ДНК-полимеразу, синтезируются дочерние молекулы ДНК.
В то время казалось вполне вероятным, что простейший механизм репликации ДНК заключается в непрерывном росте обеих новых цепей нуклеотид за нуклеотидом по мере перемещения репликационной вилки от одного конца молекулы ДНК к другому. Однако, поскольку две цепи в спирали ДНК антипараллельны, одна из дочерних цепей должна расти в направлении 5′ → 3′, а другая — в направлении 3′ → 5′. В таком случае репликационной вилке потребовалось бы две разные ДНК-полимеразы. Одна из них наращивала бы цепь в направлении 5′ → 3′ (рис. 5-34); при этом каждый поступающий мономер (дезоксирибонуклеозидтрифосфат) приносит с собой необходимую для его присоединения к цепи энергию (ее носителем является трифосфатная группа). Другая ДНК-полимераза, перемещающаяся в направлении 3′ → 5′, должна катализировать «рост с головы»; в этом случае энергию, необходимую для присоединения каждого очередного нуклеотида, должен нести конец растущей цепи ДНК. В действительности такой (3′ → 5′) ДНК-полимеразы не существует (рис. 5-38), хотя биохимикам известны некоторые другие процессы полимеризации, протекающие по типу «роста с головы» (см. рис. 2-34), Каким же образом происходит рост цепи в направлении 3′ → 5′? Возможный ответ на этот вопрос подсказали в конце 1960-х годов эксперименты с радиоактивно меченными предшественниками ДНК. Если растущие клетки получают всего на несколько секунд высокорадиоактивный 3Н-тимидин, то метка включается лишь в ДНК, синтезированную в самый последний момент, т. е. в ту ее часть, которая следует непосредственно за репликационной вилкой. Этим методом избирательного введения метки было выявлено, что при репликации бактериальной ДНК в области репликационной вилки образуются и какое-то время существуют фрагменты, насчитывающие от 1000 до 2000 нуклеотидов (впоследствии за ними закрепилось название «фрагменты Оказаки»; у эукариот они гораздо короче: от 100 до 200 нуклеотидов). Несколько позже было показано, что синтез этих фрагментов ДНК идет только в направлении 5′ → 3′; синтезированные фрагменты соединяются затем в длинные цепи ДНК под действием того же фермента, который сшивает разрывы в спирали ДНК во время ее репарации, т.е. под действием ДНК-лигазы (см. рис. 5-35).

Рис. 5-38. На первый взгляд простейшим механизмом репликации ДНК представляется механизм, изображенный на этой (неверной!) схеме. Обе дочерние цепи должны были бы при этом расти непрерывно за счет присоединения нуклеотидов соответственно в 5′ → 3′ — направлении (на рисунке — внизу) и 3′ → 5′ — направлении (на рисунке — вверху). Однако фермента, который бы катализировал присоединение нуклеотидов в направлении 3′ → 5′, не существует.

Рис. 5-39. Строение репликационной вилки. Обе дочерние цепи строятся в направлении 5′ → 3′. Для этого отстающая цепь ДНК должна синтезироваться в виде ряда коротких фрагментов (фрагменты Оказаки).
Репликационная вилка асимметрична (рис. 5-39). Из двух синтезируемых дочерних цепей ДНК одна строится непрерывной, а другая прерывистой. Первую называют ведущей (или лидирующей), а вторую — отстающей. Наращивание второй цепи отстает, потому что образование каждого фрагмента Оказаки оказывается возможным лишь после того, как продвижение ведущей цепи откроет соответствующий участок матрицы. Хотя в целом вся эта цепь строится в направлении 3′ → 5′, каждый из ее фрагментов синтезируется в направлении 5′ → 3′. Благодаря тому что ДНК на отстающей части вилки строится при помощи механизма, работающего прерывисто по типу «шитья назад иголкой» (backstitching), в репликационной вилке не требуется никакого другого фермента, кроме (5′ → 3′)-ДНК-полимеразы.
5-24
5.3.3. Высокая точность репликации ДНК предполагает наличие механизма, осуществляющего коррекцию [27]
Точность копирования при репликации ДНК столь велика, что в среднем на каждые 1-109 комплементарных пар, образующихся в процессе воспроизведения генома млекопитающих, насчитывающего 3-104 пар оснований (см. разд. 9.1.3), приходится приблизительно одна ошибка. Точность эта значительно превосходит ту, какую следует ожидать, учитывая, что во время репликации образуются не только обычные комплементарные пары оснований. В нормальной ДНК возникают на короткое время с частотой 10-4-10-5 редкие таутомерные формы всех четырех ее оснований. Эти формы образуют неправильные пары. Так, редкая таутомерная форма С спаривается с А вместо G, в результате чего возникает мутация (рис. 5-40). Таким образом высокая точность репликации ДНК определяется наличием механизмов, осуществляющих коррекцию, т. е. устраняющих подобные ошибки.

Рис. 5-40. Пример возникновения при репликации ДНК неправильной пары оснований: находясь в термодинамически невыгодной таутомерной форме, цитозин легко образует водородные связи с аденином.

Рис. 5-41. Схема, поясняющая, как протекает процесс коррекции (устранение ошибок) при синтезе ДНК, катализируемом ДНК-полимеразами у бактерий. Предполагается, что аналогичный механизм коррекции действует и в эукариотических клетках.
Один из важных механизмов коррекции зависит от особых свойств ДНК-полимеразы. В отличие от РНК-полимераз ДНК-полимеразы не могут начать синтез новой полинуклеотидной цепи, просто связав друг с другом 3′-ОН- конец какой-либо полинуклеотидной цепи, которая должна быть спарена с матричной цепью ДНК; ДНК-полимеразы способны только добавлять новые нуклеотиды к уже имеющемуся 3′-ОН-концу полинуклеотидной цепи (см. рис. 534). Эту предобразованную цепь, к которой добавляются нуклеотиды, называют затравкой или праймером. Молекулы ДНК с затравкой, у которой 3′-ОН-конец не спарен, не могут служить матрицами. Бактериальные ДНК-полимеразы способны, однако, с ними работать. Вступив в контакт с такими молекулами ДНК, они используют присущую им (3′ → 5′)-экзонуклеазную активность и отщепляют (путем гидролиза) любые неспаренные нуклеотиды на затравочном конце. Отщепляется ровно столько нуклеотидов, сколько требуется для того, чтобы у затравки появился спаренный конец и образовалась активная матрица. Действуя таким образом, ДНК-полимераза выступает в роли «самокорректирующего» фермента: она устраняет свои собственные ошибки, возникающие в процессе полимеризации. Рис. 5-41 поясняет, как этот тип коррекции может использоваться для удаления неправильных пар С—А, образуемых редкой таутомерной формой цитозина.
Потребность в правильно спаренном конце как раз и наделяет ДНК- полимеразу способностью исправлять свои собственные ошибки. Такой фермент, очевидно, мог бы начать синтез ДНК при полном отсутствии затравки, только утратив способность различать спаренный и неспаренный концы. В то же время РНК-полимеразы, участвующие в транскрипции генов (см. разд. 5.1.1), судя по всему не нуждаются в самокоррекции, потому что ошибки транскрипции не передаются следующему поколению и случайно возникшие дефектные молекулы особой роли не играют. РНК-полимеразы могут начинать синтез новых полинуклеотидных цепей в отсутствие затравки, причем ошибки встречаются с частотой 10-4 как при синтезе РНК, так и при трансляции, т.е. при переводе нуклеотидных последовательностей мРНК в аминокислотные последовательности белков.
5-22
5.3.4. Репликация ДНК в направлении 5’ → 3’ обеспечивает эффективную коррекцию
Весьма вероятно, что однонаправленность репликации ДНК (5′ → 3′) объясняется высокими требованиями к точности процесса. Если бы существовала ДНК-полимераза, присоединяющая дезоксирибонуклеозидтрифосфаты к синтезируемой полинуклеотидной цепи таким образом, что эта цепь росла в направлении 3′ → 5′, то активирующую трифосфатную группировку нес бы растущий 5′-конец цепи, а не поступающий мононуклеотид. В этом случае ошибки полимеризации не могли бы устраняться простым гидролизом, потому что появление свободного 5′-конца немедленно обрывало бы синтез ДНК. Ясно, что основание, только что неправильно спарившееся на 3′-конце, устранить гораздо легче, чем такое же основание, присоединившееся к 5′-концу цепи ДНК. Поэтому, хотя механизм репликации ДНК, изображенный на рис. 5-39, кажется на первый взгляд значительно более сложным и громоздким, чем неверный гипотетический механизм, представленный на рис. 5-38, этот реально функционирующий механизм способен обеспечить гораздо большую точность именно в силу того, что синтез ДНК идет здесь только в направлении 5′ → 3′.
5-25
5.3.5. Для синтеза коротких затравочных молекул на матрице отстающей цепи требуется особый фермент [28]
С того момента, как возникла репликационная вилка, для ДНК-полимеразы, синтезирующей ведущую цепь, всегда есть спаренный 3′-конец, необходимый ей для того, чтобы начать синтез новой цепи. Иначе обстоит дело с ДНК-полимеразой, ответственной за синтез отстающей цепи. Ей требуется всего каких-нибудь 4 с для того, чтобы синтезировать один короткий фрагмент ДНК, после чего она должна переключиться на синтез совсем другого фермента на новом участке матричной цепи, расположенной на некотором расстоянии от первого (см. рис. 5-39). Для этого ей всякий раз нужна затравка со спаренным 3′-концом, а следовательно, нужен и механизм, способный производить такие затравки. В этот механизм входит фермент, называемый ДНК-праймазой. Она синтезирует из рибонуклеозидтрифосфатов короткие РНК-затравки (праймеры), состоящие у эукариот примерно из 10 нуклеотидов (рис. 5-42). Эти затравки синтезируются с определенными интервалами на матрице для отстающей цепи; здесь их наращивает ДНК-полимераза, начиная, таким образом, всякий раз новый фрагмент Оказаки. Молекула ДНК-полимеразы продолжает это наращивание до тех пор, пока она не достигнет РНК-затравки, присоединенной к 5′-концу предыдущего фрагмента ДНК. Чтобы обеспечить образование непрерывной цепи ДНК из многих таких фрагментов, в действие вступает особая система репарации ДНК, быстро удаляющая РНК-затравку и заменяющая ее на ДНК. Завершает процесс ДНК-лигаза, соединяющая 3′-конец нового фрагмента ДНК с 5′-концом предыдущего фрагмента (рис. 5-43).
Почему предпочтение отдается удаляемой РНК-затравке, а не ДНК-затравке, которую не требовалось бы удалять? Выше мы отмечали, что самокорректирующая полимераза не способна начинать синтез полинуклеотидных цепей de novo; это предполагает и обратное утверждение: тот фермент, который начинает синтез цепей de novo, к эффективной самокоррекции не способен. Значит, любой фермент, катализирующий инициацию синтеза фрагментов Оказаки, неизбежно создал бы не слишком точную копию (не менее 1 ошибки на 105). Это означало бы колоссальное увеличение частоты мутаций даже при том, что количество таких копий, сохранившееся в конечном продукте, составляло бы не более 5% всего генома (например, 10 нуклеотидов во фрагменте, состоящем из 200 нуклеотидов). Естественно думать поэтому, что выдвижение РНК, а не ДНК на роль затравки обеспечивало важное преимущество, поскольку рибонуклеотиды автоматически метят такие последовательности, как «плохие копии», которые должны быть удалены.

Рис. 5-42. Схема реакции, катализируемой праймазой — ферментом, синтезирующим короткие РНК-затравки в отстающей цепи ДНК. В отличие от ДНК-полимеразы этот фермент способен начинать синтез новой полинуклеотидной цепи с соединения двух нуклеозидтрифосфатов. Образовав короткий полинуклеотид, праймаза прекращает работу. Теперь к свободному 3′-концу может добавлять нуклеотиды ДНК-полимераза.
5-26
5-33
5.3.6. Особые белки способствуют расплетанию двойной спирали ДНК перед репликационной вилкой [29]
Двойная спираль ДНК должна расплетаться по ходу продвижения репликационной вилки, для того чтобы поступающие дезоксирибонуклеозидтрифосфаты могли спариваться с родительской матричной цепью. Однако в обычных условиях двойная спираль ДНК весьма стабильна; спаренные основания соединены столь прочно, что для разделения двух цепей ДНК в пробирке требуются температуры, приближающиеся к точке кипения воды (90°С). По этой причине большинство ДНК-полимераз может копировать лишь ту молекулу ДНК, у которой матричная цепь уже отделилась от другой цепи. Для того чтобы двойная спираль ДНК раскрылась и соответствующая матричная цепь стала доступной для ДНК-полимеразы, необходимы особые белки. Они бывают двух типов.
ДНК-геликазы были впервые выделены как белки, которые, присоединяясь к одиночной цепи ДНК, катализируют гидролиз АТР. Как уже отмечалось в гл. 3, гидролиз АТР может циклическим образом изменять форму молекулы белка, вследствие чего белок будет производить механическую работу (см. разд. 3.4.11). Именно этот принцип лежит в основе быстрого перемещения ДНК-геликаз по одиночной цепи ДНК. Встречая на своем пути участок двойной спирали, эти ферменты продолжают двигаться вдоль своей цепи и тем самым расплетают двойную спираль (рис. 5-44). Расплетание ДНК-спирали в области репликационной вилки, вероятно, осуществляется двумя совместно действующими ДНК- геликазами, одна из которых перемещается по ведущей цепи, а другая — по отстающей. Ясно, что две эти геликазы должны двигаться вдоль одиночных цепей ДНК в противоположных направлениях, т. е. это должны быть разные ферменты. Действительно, оба указанных типа ДНК- геликаз удалось обнаружить. При этом исследования на бактериях показали, что главную роль играет ДНК-геликаза отстающей цепи. Причины этого мы обсудим ниже.
Белки, дестабилизирующие спираль (их называют также белками, связывающими одноцепочечную ДНК или SSB-белками), связываются с одиночными цепями ДНК, не закрывая оснований, т. е. оставляя их доступными для спаривания. Сами они не способны расплетать длинные молекулы ДНК, но, присоединяясь к одиночным цепям ДНК, они тем самым способствуют любому процессу расплетания спирали; они, например, помогают ДНК-геликазе расплетать двойную спираль в репликационной вилке. На матрице отстающей цепи SSB-белки кооперативным образом связываются с одноцепочечными участками ДНК и предотвращают здесь образование «шпилек», небольших двухспиральных структур, которые могли бы помешать синтезу ДНК, осуществляемому ДНК-полимеразой (рис. 5-45).

Рис. 5-43. Отдельные этапы синтеза каждого из фрагментов отстающей цепи ДНК. У эукариот РНК-затравки синтезируются в отстающей цепи с интервалами приблизительно в 200 нуклеотидов и каждая из них состоит из 10 нуклеотидов.
5.3.7. Белки в репликационной вилке действуют кооперативно, образуя «репликационную машину» [30]
До сих пор мы говорили о репликации ДНК так, как если бы она осуществлялась смесью репликационных белков, действующих независимо друг от друга. Между тем в действительности большая часть этих белков объединена в крупный мультиферментный комплекс, быстро движущийся вдоль ДНК. Этот комплекс — нечто вроде крошечной «швейной машины»: «деталями» его служат отдельные белки, а источником энергии — реакция гидролиза нуклеозидтрифосфата. Комплекс изучен достаточно хорошо только у бактерий Е. coli и у некоторых вирусов, но есть все основания считать, что очень похожий механизм действует и у эукариот (см. разд. 9.3.3).
Схема на рис. 5-46, где подробно изображена репликационная вилка, позволяет судить о том, как работают отдельные части такой «репликационной машины». В области вилки действуют две идентичные ДНК-полимеразы — на ведущей и на отстающей цепи. Спираль ДНК расплетается в результате совместного действия ДНК-полимеразы, работающей на ведущей цепи, и ДНК-геликазы, движущейся вдоль отстающей цепи; этому процессу способствуют кооперативно связывающиеся молекулы дестабилизирующегося белка. В то время как на ведущей цепи ДНК- полимераза работает непрерывно, на отстающей цепи фермент через определенные интервалы прерывает и вновь возобновляет свою работу, используя для полимеризации короткие РНК-затравки, синтезируемые ДНК-праймазой.
Эффективность репликации сильно возрастает вследствие тесного объединения всех этих белковых компонентов. Молекула праймазы непосредственно сцеплена с ДНК-геликазой, образуя вместе с нею на отстающей цепи структуру, называемую праймосомой, которая движется с репликационной вилкой и по ходу своего движения синтезирует РНК-затравки. Молекула ДНК-полимеразы, работающая на отстающей цепи, также движется совместно с остальными белками, синтезируя ряд новых фрагментов Оказаки; ради этого, как полагают, цепь ДНК, которая служит для нее матрицей, складывается сама на себя, как это показано на рис. 5-47. Репликационные вилки оказываются, таким образом, объединены в одну крупную структуру (с общей массой > 106 дальтон), быстро перемещающуюся вдоль ДНК и обеспечивающую возможность координированного и эффективного синтеза ДНК на обет ветвях вилки.

Рис. 5-44. Действие ДНК-геликаз. Небольшой фрагмент ДНК присоединен путем отжига к длинной одноцепочечной ДНК, так что образовался короткий участок двойной спирали. Эта спираль расплетается по мере того, как геликаза движется вдоль одиночной цепи ДНК, катализируя реакцию, для которой требуется наряду с ферментом и АТР. Источником энергии для движения геликазы служит гидролиз АТР (см. рис. 3-63).

Рис. 5-45. Влияние дестабилизирующих белков на структуру одно-цепочечной ДНК. Поскольку каждая белковая молекула предпочитает связываться с другой, уже связавшейся ранее молекулой (так называемое кооперативное связывание), эти белки образуют длинные кластеры, выпрямляющие матричные пени ДНК и облегчающие процесс полимеризации. Структуры в форме «шпильки», возникающие в свободной одноцепочечной ДНК, образуются путем случайного спаривания оснований в коротких участках, содержащих взаимно комплементарные последовательности нуклеотидов, они напоминают короткие спирали, возникающие во всех молекулах РНК.

Рис. 5-46. Главные типы белков, действующих в области репликационной вилки (схема показывает их локализацию на ДНК). Комплекс ДНК-праймазы и ДНК-геликазы на отстающей цепи ДНК известен под названием праймосомы.
Позади «репликационной машины» по ходу ее движения остается на отстающей цепи ряд несшитых фрагментов Оказаки, все еще содержащих на своем 5′-конце РНК-затравки, необходимые для инициации на синтеза. Эти РНК-затравки должны быть удалены, а фрагменты сшиты при помощи репарирующих ферментов, работающих позади репликационной вилки (см. рис. 5-43).

Рис. 5-47. Схема, иллюстрирующая современные представления о расположении репликационных белков в движущейся репликационной вилке. Вместо двумерной структуры, изображенной на рис. 5-46, здесь показано, как ДНК на отстающей цепи складывается, в результате чего возникает комплекс из двух ДНК-полимераз — ведущей и отстающей цепи. Кроме того, благодаря складыванию 3′-конец каждого завершенного фрагмента Оказаки оказывается рядом со стартовым участком следующего такого фрагмента (ср. с рис. 5-46). Находясь в тесном контакте с остальными репликационными белками, молекула ДНК-полимеразы отстающей цепи может непрерывно работать на одной и той же репликационной вилке, отделяясь от готового фрагмента ДНК, она переходит к ближайшей новой РНК-затравке, чтобы начать синтез следующего фрагмента. Обратите внимание, что на этой схеме одна из дочерних спиралей ДНК направлена вправо и вниз, а вторая влево и вверх.
5.3.8. Ошибки при репликации ДНК в бактериальных клетках устраняются особой корректирующей системой, распознающей неправильное спаривание оснований
У таких бактерий, как Е. coli, деление происходит каждые 30 мин, поэтому у них сравнительно легко выявить в большой популяции клеток редкие экземпляры с измененными признаками. Выделен, например, класс мутантов, характеризующихся резким повышением частоты спонтанных мутаций, что связано с присутствием в его клетках специфических генов-мутаторов. Известен ген-мутатор, кодирующий дефектную форму 3′ → 5′-корректирующей экзонуклеазы, представляющей собой субъединицу ДНК-полимеразы (см. разд. 5.3.3). Если дефект затрагивает этот белок, то ДНК-полимераза утрачивает способность эффективно осуществлять коррекцию и в ДНК накапливается много ошибок, которые при нормальной репликации были бы устранены.
Изучение тех мутантов Е. coli, у которых имеются гены-мутаторы, выявило еще одну систему, в норме устраняющую ошибки репликации, не улавливаемые корректирующей экзонуклеазой. Эта система коррекции неправильного спаривания (mismatch proofreading sistem), называемая также системой исправления ошибок спаривания (mismatch repair system), отличается от ранее рассмотренных систем репарации ДНК тем, что она не зависит от присутствия в ДНК аномальных нуклеотидов, которые должны быть распознаны и удалены («вырезаны»). Она выявляет деформации на внешней стороне спирали, вызванные плохой пригонкой обычных, но некомплементарных оснований. Если бы эта корректирующая система просто распознавала ошибки спаривания в реплицировавшейся ДНК и удаляла без выбора один из двух неправильно спарившихся нуклеотидов, то в половине случаев она бы сама совершала ошибку, «исправляя» не новосинтезированную, а матричную цепь, так что в среднем частота ошибок оставалась бы прежней. Для эффективной коррекции система должна уметь различать неправильно спаривающиеся нуклеотиды и избирательно удалять такие нуклеотиды только из новой цепи (т.е. устранять именно ошибки репликации).
В клетках Е. coli процесс распознавания связан с метилированием определенных остатков аденина в ДНК. Метальные группы присоединяются ко всем остаткам А в последовательности GATC, но лишь спустя некоторое время после того, как А включится в новосинтезированную цепь ДНК. Новые цепи отличаются от старых тем, что только в них сразу же за репликационной вилкой могут находиться еще не метилированные последовательности GATC. Коррекция неправильного спаривания осуществляется крупным мультиферментным комплексом, сканирующим каждую из двух цепей двойной спирали ДНК. Этот комплекс удаляет только неправильно присоединенные нуклеотиды, но делает это лишь после того, как на той же цепи обнаружится и неметилированная последовательность GATC. Поэтому нуклеотиды удаляются только из новой цепи, т. е. устраняются ошибки репликации (рис. 5-48).

Рис. 5-48. Схема эксперимента, иллюстрирующего работу системы коррекции неправильного спаривания, устраняющей у бактерий ошибки репликации ДНК. Особый белковый комплекс удаляет неспаренные нуклеотиды из вновь синтезируемой цепи ДНК позади репликационной вилки, этот репарирующий комплекс узнает новую цепь ДНК по обнаруживаемым в ней неметилированным последовательностям GATC. На схеме представлены три молекулы ДНК с одной и той же «неправильной» парой нуклеотидов, но при этом в одной молекуле (А) метилированные последовательности GATC встречаются в обеих цепях, в другой молекуле (Б) таких метилированных последовательностей нет совсем, а в третьей (В) они присутствуют только в одной из цепей. Если воздействовать на эти молекулы ДНК клеточным экстрактом, содержащим корректирующий комплекс, то мы получим представленный здесь результат. Молекула ДНК в правой части рисунка воспроизводит картину, обнаруживаемую непосредственно за репликационной вилкой: нижняя цепь соответствует новой цепи, где метилирование еще не произошло.
В эукариотических клетках не удалось пока выявить ни одного из этих двух механизмов коррекции, обнаруженных у бактерий. Однако степень точности репликации у млекопитающих и у Е. coli приблизительно одинакова, и потому можно думать, что оба описанных типа коррекции существуют и у эукариот. Следует, впрочем, отметить, что в ДНК млекопитающих нет метилированных остатков А, поэтому механизм, который используется системой репарации ошибок спаривания для узнавания новосинтезированной цепи, должен быть в данном случае иным.
5-27
5.3.9. Репликационные вилки возникают в точках начала репликации [32]
И у бактерий, и у млекопитающих образование репликационных вилок начинается с возникновения особой структуры, называемой репликационным глазком (replication bubble). Это небольшой участок, в котором две цепи родительской спирали ДНК отделились одна от другой и были использованы в качестве матриц для синтеза ДНК (рис. 5-49). Для бактерий и некоторых вирусов, размножающихся в эукариотических клетках, удалось показать, что репликационный глазок образуется в тех местах молекулы ДНК, где находятся специфические нуклеотидные последовательности, получившие название точек начала репликации. Эти последовательности состоят приблизительно из 300 нуклеотидов. Предполагают, что аналогичные точки начала репликации существуют и в эукариотических хромосомах, однако надежных доказательств этого пока нет (см. разд. 9.3.2).
Процесс возникновения репликационных вилок удалось в некоторых случаях воспроизвести in vitro. Эти опыты показали, что у бактерий и бактериофагов инициация репликационных вилок начинается так, как это представлено на рис. 5-50. Множество копий инициаторного белка связываются с особыми участками в точке начала репликации, образуя крупный белковый комплекс. Этот комплекс присоединяет затем ДНК-геликазу и помещает ее на свободную одиночную цепь ДНК в прилегающем участке спирали. Присоединяется также ДНК-праймаза, т. е. образуется праймосома, которая, двигаясь от точки начала репликации, синтезирует РНК-затравку, что дает возможность начать синтез первой цепи ДНК. Остальные белки быстро объединяются после этого в два репликационных белковых комплекса, которые теперь движутся от точки начала репликации в противоположных направлениях (см. рис. 5-49); они продолжают синтезировать ДНК до тех пор, пока обе вилки не пройдут путь по матрице до самого конца.
Некоторые дополнительные данные, касающиеся инициации репликационных вилок в хромосомах эукариот, мы обсудим в гл. 9, там, где речь пойдет о клеточном ядре.

Рис. 5-49. Гипотетический механизм образования репликационных вилок в точках начала репликации (см. также рис. 5-50).

Рис. 5-50. Упрощенная схема, иллюстрирующая начальные этапы образования репликационных вилок в точках начала репликации у Е. coli и бактериофага X. Для обнаружения данного механизма потребовались опыты in vitro с использованием смеси высокоочищенных белков.
Последующие этапы приводят (пока не ясным путем) к инициации еще трех цепей ДНК (рис. 5-49). У Е. coli в репликации ДНК роль инициаторного белка играет белок dnaA; а праймосома состоит из белков dnaB (ДНК-геликаза) и dnaG (ДНК-праймаза).
5.3.10. ДНК-топоизомеразы предотвращают спутывание ДНК во время репликации [33]
Изображая спираль ДНК так, как мы это делали до сих пор, т.е. неправильно, в виде плоской «лестницы», мы игнорировали «проблему закручивания» (winding problem). Между тем на каждые 10 пар оснований, образующихся в репликационной вилке, родительская двойная спираль должна совершить один полный оборот вокруг своей оси. Следовательно, для того чтобы репликационная вилка могла продвигаться вперед, вся хромосома впереди нее должна быстро вращаться (рис. 5-51), что для длинных хромосом потребовало бы большой затраты энергии. При репликации ДНК эта проблема решается иначе: путем образования в спирали своего рода «шарнира», особого класса белков, называемых ДНК- топоизомеразами.
ДНК-топоизомераза представляет собой нечто вроде «обратимой нуклеазы». Сначала она разрывает цепь ДНК, а затем ковалентно присоединяется к разорванному концу. Ковалентная связь белок — ДНК обладает довольно значительной энергией, потому что в ней сохраняется энергия разорванной фосфодиэфирной связи. Вследствие этого реакция, приводящая к разрыву цепи, обратима и не требует дополнительной затраты энергии. В этом отношении данный механизм существенно отличается от механизма действия ДНК-лигазы, о котором мы говорили выше (см. рис. 5-35).
Существуют различные типы ДНК-топоизомераз. Топоизомераза типа I разрывает только одну из двух цепей двойной спирали ДНК, что дает возможность двум участкам ДНК по обе стороны от разрыва свободно вращаться относительно друг друга вокруг фосфодиэфирной связи, находящейся напротив разрыва, которая в этом случае выполняет роль упомянутого выше «шарнира» (рис. 5-52). Всякое напряжение в спирали ДНК заставляет ее вращаться в таком направлении, чтобы ослабить это напряжение. Поэтому вращение во время репликации ДНК происходит лишь на коротком отрезке спирали — в той части, которая находится непосредственно перед репликационной вилкой. Аналогичная проблема, возникшая в процессе транскрипции ДНК, решается таким же путем.

Рис. 5-51. «Проблема кручения», возникающая при репликации ДНК. Для того чтобы репликационная вилка (у бактерий) могла продвигаться вперед со скоростью 500 нуклеотидов в 1 с, родительская спираль ДНК перед вилкой должна вращаться со скоростью 50 об/с.

Рис. 5-52. Обратимая реакция, приводящая к появлению разрыва в одной из цепей ДНК. Реакция у эукариот катализируется ДНК-топоизомеразой типа I Ферменты этой группы образуют временную ковалентную связь с ДНК.

Рис. 5-53. Пример реакции разделения двух сцепленных кольцевых молекул ДНК, катализируемой ДНК-топоизомеразой типа II. Действие этих ферментов (в отличие от реакций, катализируемых ДНК-топоизомеразами типа I) сопряжено с гидролизом АТР и некоторые из них способны сообщать спирали ДНК дополнительное напряжение. ДНК-топоизомеразы типа II обнаруживаются и у прокариот, и у эукариот, по всей вероятности, они участвуют во многих реакциях, имеющих отношение к ДНК.
Топоизомераза типа II ковалентно связывается с обеими цепями двойной спирали ДНК и вносит в нее на время двухцепочечный разрыв. Ферменты этого типа активируются под действием тех участков на хромосомах, где перекрестились спирали. Присоединившись к такому перекресту, топоизомераза: 1) обратимо разрывает одну из двух двойных спиралей, создавая тем самым для другой своего рода «ворота», 2) вынуждает вторую двойную спираль пройти через этот разрыв и 3) сшивает обе разорванные цепи, а затем отделяется от ДНК. Действуя подобным образом, топоизомеразы типа II очень быстро разделяют две сцепленные кольцевые молекулы ДНК (рис. 5-53). Точно так же предотвращают они и спутывание молекул ДНК, которое в противном случае неизбежно создавало бы при репликации серьезную проблему. Известны температурочувствительные мутанты дрожжей, вырабатывающие топоизомеразу II, которая при 37°С инактивируется. Если нагреть эти дрожжевые клетки до такой температуры, то их хромосомы в процессе митоза остаются спутанными и не могут разойтись. Насколько необходим для распутывания хромосом такой «инструмент», как топоизомераза II, поймет каждый, кто хоть раз пытался распутать безнадежно запутавшуюся леску, не имея под рукой ножниц.
5-28
5.3.11. Репликация ДНК у эукариот и прокариот в основных чертах сходна [24]
Почти все, что мы знаем о репликации ДНК, удалось выяснить в опытах с очищенными мультиферментными системами бактерий и бактериофагов, способными осуществлять репликацию ДНК in vitro. Получение таких систем в 1970-х годах заметно облегчилось после того, как удалось выделить мутанты по целому ряду различных генов, ответственных за репликацию, которые можно было использовать для идентификации и очистки соответствующих белков (рис. 5-54).
У эукариот энзимология репликации ДНК пока еще детально не изучена, главным образом потому, что получать здесь необходимые мутантные формы гораздо труднее. Однако схема репликации у прокариот и эукариот в основных чертах, включая геометрию репликационной вилки и потребность в РНК-затравке, по-видимому, одинакова. Главное различие заключается в том, что у эукариот ДНК реплицируется не как таковая, а в виде хроматина, в котором она прочно связана с белками, принадлежащими к классу гистонов. В гл. 8 мы узнаем, что гистоны образуют комплексы в форме дисков, вокруг которых обвивается эукариотическая ДНК, в результате чего возникают регулярно повторяющиеся структуры, называемые нуклеосомами. Нуклеосомы располагаются вдоль молекулы ДНК с интервалами 200 пар оснований. Быть может, именно этим объясняется тот факт, что новые фрагменты отстающей цепи ДНК закладываются у эукариот с интервалами в 10 раз более короткими (от 100 до 200 нуклеотидов), чем у бактерий (от 1000 до 2000 нуклеотидов). Кроме того, если нуклеотиды служат барьерами, на время останавливающими продвижение ДНК-полимеразы, присутствие хроматина (а не голой ДНК) может, вероятно, объяснить и то, что репликационные вилки движутся у эукариот приблизительно в 10 раз медленнее, чем у бактерий.

Рис. 5-54. Получение у бактерий и бактериофагов мутантов с различными нарушениями репликации ДНК открыло возможности для выявления и очистки ферментов, выполняющих какую-либо еще не известную функцию, необходимую для репликации ДНК у прокариот. Использованные здесь температурочувствительные мутанты принадлежат к так называемым условным мутантам, обычно их фермент нормально функционирует при низкой температуре и не работает при высокой. У «безусловных» мутантов с нарушениями репликации синтез ДНК не идет ни при низкой, ни при высокой температуре, и потому эти мутанты обречены на гибель. В модифицированной форме такие «тесты на комплементацию in vitro» полезны также при биохимическом изучении многих других процессов.
Заключение
Самокорректирующая ДНК-полимераза катализирует полимеризацию нуклеотидов на обеих цепях спирали ДНК в направлении 5′ → 3′, копируя матрицу с высокой степенью точности. Поскольку две цепи двойной спирали ДНК аптипараллелъны, в направлении 5′ → 3′ может непрерывно синтезироваться лишь одна из двух цепей (ее называют ведущей). Другая, отстающая цепь синтезируется в виде коротких фрагментов по принципу «шитья назад иголкой». Самокорректирующая ДНК-полимераза не способна начинать синтез новой цепи. Поэтому для закладки фрагментов отстающей цепи ДНК используются короткие молекулы РНК-затравки, которые позже удаляются — их заменяет ДНК.
Процесс репликации ДНК требует совместного действия многих белков. В нем участвуют: 1) ДНК-полимераза и ДНК-праймаза, катализирующие полимеризацию нуклеозидтрифосфатов; 2) ДНК-геликазы и дестабилизирующие белки, помогающие расплести спираль ДНК, которую предстоит копировать; 3) ДНК-лигаза и фермент, разрушающий молекулы РНК-затравки; они нужны для сшивания прерывисто синтезируемых фрагментов отстающей цепи ДНК; 4) ДНК-топоизомеразы, помогающие решить проблемы кручения и спутывания спирали ДНК; 5) инициаторные белки, присоединившиеся к специфическим последовательностям ДНК в точке начала репликации и способствующие образованию здесь новой репликационной вилки. В точке начала репликации к ДНК-матрице сначала присоединяется белковый комплекс, состоящий из ДНК- геликазы и ДНК-праймазы (его называют праймосомой); затем к этому комплексу добавляются другие белки и возникает мультиферментный комплекс — «репликационная машина», которая и осуществляет синтез ДНК.
Транскрипционные факторы не только регулируют работу генов, но могут обладать и другими, порой неожиданными и даже нежелательными функциями. В этой статье — рассказ о такой функции, когда транскрипционный фактор C/EBP крепко связывается с частично поврежденной ДНК и не позволяет системе репарации починить спрятанный под собой участок ДНК. А что он спрятал и почему — сейчас и расскажем!
Долгое время было принято считать, что самую важную роль в геноме, то есть в ДНК, играют только белок-кодирующие участки — гены, кодирующие последовательности белков. Которые, к слову, у человека занимают лишь 1–2% от длины генома [1], [2]. Остальную часть генома называли темной материей, или мусорной ДНК, — разумеется, ошибочно. Но сегодня научные исследования, посвященные некодирующим участкам ДНК и их функциональной роли, расцветают все ярче и ярче.
Действительно, некодирующая ДНК способна на многое.
Первое, о чем обычно вспоминают, — повторяющиеся последовательности [3]. Правда, полезных их функций нам известно маловато.
Затем — гены некодирующих РНК, которые самостоятельно или при участии белков выполняют различную работу в клетке [4]. Систематическим изучением таких РНК занимается, например, международный консорциум FANTOM6.
Но и помимо повторов и РНК-генов, некодирующие участки генома содержат много интересного. Например, в них находятся регуляторные элементы (промоторы и энхансеры), которые определяют активность генов — как кодирующих, так и не кодирующих белки, — в различных типах клеток и в разных условиях. Работают они за счет привлечения специальных белков — факторов транскрипции, — которые способны распознать в ДНК сравнительно короткие, но характерные, паттерны (мотивы). В пределах регуляторных районов генома эти факторы связывают подходящие участки ДНК, а затем образуют белок-белковые комплексы друг с другом и с компонентами молекулярной машины, осуществляющей транскрипцию генов, — то есть непосредственно синтез РНК. Именно участками посадки факторов транскрипции (так называемыми ДНК-сайтами связывания, или просто сайтами связывания) мы, то есть наш дружный коллектив соавторов теперь уже опубликованной статьи [5], занимаемся уже больше 10 лет. Кстати, о компьютерном предсказании сайтов связывания в масштабе полного генома (на основе нашей работы) можно прочитать в статье «Биомолекулы» «Мечту вызывали?» [6].
Участки ДНК, непосредственно взаимодействующие с факторами транскрипции в геноме человека, сравнительно коротки — 10–20 нуклеотидов. Сила взаимодействия фактора транскрипции с ДНК (и как следствие, активность зависимого от него гена) может существенно меняться даже при замене всего одного нуклеотида-«буквы» (например, это приводит к аллель-специфичному связыванию участков [7], различающихся между материнской и отцовской хромосомами). То есть даже однонуклеотидные мутации могут вызывать серьезные нарушения генной регуляции: влиять на судьбу клеток и осложнять течение заболеваний. В частности, такие мутации способны превращать стволовые клетки человека в опухолевые или приводить к болезням, связанным с неправильной дифференцировкой клеток. Во многих опухолях часто находят специфичные мутации в промоторе гена теломеразы TERT [8]. Этот фермент необходим для репликации концов хромосом во время деления клеток. Логично, что в соматических клетках, которые не делятся, ген теломеразы находится в неактивном состоянии. Но если в промоторе этого гена происходят указанные выше специфичные мутации, то факторы транскрипции вновь начинают связываться с промотором, тем самым активируя ген теломеразы. Это позволяет клетке активно делиться, а эта способность, как мы знаем, — важное свойство опухолевых клеток [9].
Мутации, происходящие в соматических клетках (то есть во всех, кроме половых), называют соматическими. Обычно их изучают в опухолевых клетках, где часто затруднена работа систем репарации, а потому мутации накапливаются с высокой скоростью. На возникновение и частоту мутаций в популяции раковых клеток влияет сразу множество факторов: и отбор (как положительный, так и отрицательный), и активность процессов, напрямую вызывающих повреждения ДНК, и различная эффективность систем репарации. В общем, целый клубок, который достаточно сложно расплести, а результаты анализа, в итоге, сложно однозначно интерпретировать. Мы решили зайти с другой стороны и использовали опубликованные данные по мутагенезу соматических стволовых клеток взрослых и здоровых людей, где основной вклад в накопление мутаций вносят непосредственно мутационные процессы, без поломки репарационных систем и следов естественного отбора.
Для начала мы разделили мутации на группы, соответствующие известным мутационным процессам: например, вызванные влиянием активных форм кислорода или дезаминированием 5-метилцитозина. Затем мы наложили координаты мутаций на геномную карту всех известных участков связывания транскрипционных факторов. Оказалось, что как минимум треть мутаций расположена недалеко от участков связывания. Теперь имело смысл внимательно посмотреть на районы работы конкретных факторов транскрипции по отдельности.
Мы сопоставили паттерны в ДНК, узнаваемые определенными факторами транскрипции, с точным положением мутаций в пределах небольших «окошек» в геноме (не далее 50 нуклеотидов от мутации). Выяснилось, что мутации CpG-динуклеотидов вида [C>T]G, вызываемые дезаминированием 5-метилцитозина, неожиданно часто попадают точно в центр участков связывания белков C/EBP. Интересно, что этот эффект ранее обнаруживали для опухолевых клеток [10], но не смогли интерпретировать.
C/EPB: что за зверь?
Факторы транскрипции семейства C/EBP (CCAAT/enhancer-binding proteins) необходимы для нормальной дифференцировки различных типов клеток. Они активируют различные гены, взаимодействуя с их промоторами, а также могут взаимодействовать с белками клеточного цикла. Интересно, что белки этого семейства могут как подавлять переход соматических клеток в опухолевые, так и активировать его [11].
Чаще всего метилирование ДНК осуществляется по цитозину в динуклеотидах CpG. Это она из наиболее распространенных эпигенетических модификаций . Повышенная частота мутаций метилированных CpG-динуклеотидов за счет дезаминирования метилцитозина известна очень давно, но почему же участки связывания C/EBP мутируют еще в 2–4 раза чаще, чем ожидается? Могут ли транскрипционные факторы напрямую провоцировать дезаминирование цитозина в метилированных CpG в пределах своих сайтов? Эта гипотеза первой приходит в голову, но, сразу скажем, она слишком фантастическая. Ведь прочное связывание фактора транскрипции, наоборот, должно бы приводить к дополнительной защите ДНК от изменений.
Более обстоятельно с эпигенетикой вы можете познакомиться в одноименном спецпроекте «Биомолекулы», — в частности, в статье «Молекулы и эпигеном» [12].
Второй вариант, который мы рассматривали, был такой: может ли быть так, что транскрипционный фактор не дает возможности репарационной машине добраться до неспаренного основания и починить его? Например, из-за избыточно крепкой связи с таким «несовершенным» участком двойной спирали. Эта возможность — прочное связывание конкретных вариантов сайтов с неспаренными основаниями — недавно была показана для многих транскрипционных факторов. Но не для белков C/EBP, что тоже наталкивало на сомнения в «работоспособности» предположения, но мы решили его проверить.
Беспечный ездок
Один из широкого изучаемых белков семейства C/EBP — C/EBPβ. Для него в банке данных PDB доступны закристаллизованные комплексы ДНК-связывающего домена с каноническим (наилучшим возможным) ДНК-сайтом. За помощью мы обратились к коллегам (а они, забегая вперед в нашем рассказе, стали соавторами этой статьи), которые на основе этих структур провели моделирование комплекса белка C/EBPβ с различными вариантами участков ДНК. В одном случае, вместо цитозина в CpG-пару по одной из цепей вводили тимин, оставляя гуанин на противоположной цепи (тем самым образуя неверно спаренные нуклеотиды; см. рис. 1 для подробностей). А в другом случае вместе с заменой цитозина на тимин поменяли еще и гуанин на аденин, чтобы была сформирована каноническая пара — такая, какой она будет в одной из дочерних клеток после репликации ДНК и деления клетки с мутацией. И вот что интересно: наибольшее сродство к белку было у фрагмента ДНК, содержащего неканоническое спаривание нуклеотидов!
![Предложенная модель фиксации мутаций ([C>T]G)](https://biomolecula.ru/img/content/3187/3187-01.model-fiksacii-mutacij.png "Предложенная модель фиксации мутаций ([C>T]G)")
Рисунок 1. Предложенная модель фиксации мутаций ([C>T]G), вызванных дезаминированием метилцитозина и повышенным сродством белков C/EBP к «поврежденным» сайтам в ДНК. А сродство белка к ДНК повышается благодаря одноцепочечной замене в центре сайта связывания.
Выходит, идея о том, что транскрипционный фактор мешает системе репарации, действительно может работать! Но расскажем о структурном моделировании до конца: если ранжировать по силе связи, то после этой последовательности (с неидеальной спиралью и наибольшим сродством к C/EBPβ-белку) шла каноническая форма, и только затем — та, где цитозин мутировал в тимин, а гуанин — в аденин. То есть и каноничный комплекс со спариванием G·C, и комплекс после мутации и репликации с A·T, не могут тягаться по силе с тем, как тяготеет транскрипционный фактор к неспаренным основаниям G·T. Здесь интересно, что мутировавший сайт c TG-, вместо CG-динуклеотида, существенно хуже связывает C/EBP. То есть в отсутствие давления естественного отбора геном мог бы постепенно лишиться всех сильных сайтов C/EBP. Судя по всему, сайты действительно постепенно теряются, но отбор «старается» сохранить полноценный сайт хотя бы на одной из парных хромосом, ведь гетерозиготные сайты C/EBP часто являются именно парой из CG- и TG-сайтов, как мы видели в работе [5].
Такая неожиданная согласованность результатов, пришедших и из статистического анализа мутаций, и из структурного моделирования, мотивировала нас проверить, как поведет себя комплекс в реальном мире. Ведь хотелось не только моделей, но и эксперимента.
Поэтому мы провели анализ электрофоретической подвижности в геле методом EMSA для точно таких же фрагментов ДНК, ровно как и в структурном моделировании (канонический участок связывания, фрагмент с неверно спаренными G·T-нуклеотидами и фрагмент «после репликации» со спариванием A·T). И эксперимент EMSA подтвердил высокое сродство комплекса именно с вариантом с неверно спаренными нуклеотидами.
В этом эксперименте мы «следили» за радиоактивно меченым фрагментом ДНК, в котором участок связывания был каноническим. Когда формируется комплекс с белком, подвижность ДНК в геле замедляется. Чтобы узнать о том, как мутации влияют на связывание C/EBPβ, и как при этом изменяется подвижность в геле, была устроена конкуренция между радиоактивно меченым каноническим (видимым) и немеченными (невидимыми) участками связывания ДНК. То есть мы устроили соревнование между каноническим участком связывания, фрагментом с неверно спаренными G·T-нуклеотидами и фрагментом «после репликации» со спариванием A·T. И что же получилось? А получилось, что победил в соревновании фрагмент ДНК с мутацией [C>T]pG! Он вытеснял белок C/EBPβ из комплекса даже лучше, чем канонический участок связывания!
Таким образом, мы сначала предсказали, а затем подтвердили — C/EBP чрезвычайно прочно связывает сайты ДНК после спонтанного дезаминирования метилцитозина в тимин. И, соответственно, система репарации не может добраться до замены и восстановить правильное их спаривание. А модель закрепления мутаций и общее описание проделанной работы можно изучить на рис. 1 и 2.

Рисунок 2. Краткое визуальное описание результата исследования
Печальная и поучительная история о конкуренции в науке
И, в завершение, короткая и немного грустная история о первенстве и конкуренции в науке. Год назад мы опубликовали препринт этой работы [13] на сервере bioRxiv. Затем статья некоторое время искала «свой журнал», а потом довольно долго находилась на рецензии в Cell Reports. И вроде бы, по меркам нашей науки, не так много времени ушло на рецензирование и ответы редактора, но пару месяцев назад, уже на финальной стадии, мы неожиданно обнаружили — вышла аналогичная работа в журнале Nucleic Acids Research, в которой показано примерно то же самое [14]. Причем на сложный путь от первоначальной подачи статьи в журнал до итоговой публикации авторам потребовалось всего лишь полтора месяца!
Совпадение? В научном мире в популярной теме, можно сказать, находящейся «на передовой», это не редкость. Может быть, авторы прочитали, но почему-то не стали цитировать наш прошлогодний препринт? Может быть, статью опубликовали так быстро, потому что один из авторов еще и редактор этого журнала? Можем ли мы вообще называть себя «первооткрывателями» интересного феномена? Ведь до нас успели опубликовать практически идентичные результаты, а наша статья все еще находилась на рассмотрении. К счастью, после связи с редакцией Cell Reports процесс публикации нашей статьи ускорился, и вот, 8 июня 2021 года, и наша статья [5], наконец, вышла.
Стоит ли возмущаться такой ситуацией, стоит ли винить рецензентов, которые незлонамеренно затягивали рецензирование? Является ли опубликованный в открытом доступе препринт гарантией научного первенства? Мы не знаем ответов на эти вопросы, но сама история может быть любопытной для тех, кому интересны разные грани работы ученых в современном мире, даже разочаровывающие. Но в науке, как и везде, где есть конкуренция, такие истории всегда будут случаться, и самое главное — не переживать и продолжать копать что-нибудь интересное.
Авторы выражают благодарность Марине Фридман и Алле Федоровой за помощь в работе над текстом.
- Геном человека: полезная книга, или глянцевый журнал?;
- Сколько сора в нашей ДНК;
- Повтор, еще повтор!;
- Обо всех РНК на свете, больших и малых;
- Anna S. Ershova, Irina A. Eliseeva, Oleg S. Nikonov, Alla D. Fedorova, Ilya E. Vorontsov, et. al.. (2021). Enhanced C/EBP binding to G·T mismatches facilitates fixation of CpG mutations in cancer and adult stem cells. Cell Reports. 35, 109221;
- Мечту вызывали?;
- Мухина В. (2021). Аллели регуляторных областей ДНК повлияли на поведение транскрипционных факторов. N+1;
- Rebecca C Hennessey, Kevin M Brown. (2021). Cancer regulatory variation. Current Opinion in Genetics & Development. 66, 41-49;
- François Hafezi, Danielle Perez Bercoff. (2020). The Solo Play of TERT Promoter Mutations. Cells. 9, 749;
- Collin Melton, Jason A Reuter, Damek V Spacek, Michael Snyder. (2015). Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat Genet. 47, 710-716;
- Manlio Tolomeo, Stefania Grimaudo. (2020). The “Janus” Role of C/EBPs Family Members in Cancer Progression. IJMS. 21, 4308;
- Молекулы и эпигеном ;
- Ershova A.S., Eliseeva I.A., Nikonov O.S., Fedorova A.D. et al. (2020). Enhanced C/EBPs binding to C>T mismatches facilitates fixation of CpG mutations. bioRxiv;
- Jie Yang, John R Horton, Kadir C Akdemir, Jia Li, Yun Huang, et. al.. (2021). Preferential CEBP binding to T:G mismatches and increased C-to-T human somatic mutations. Nucleic Acids Research. 49, 5084-5094.
При транскрипции, так же как и при любом
матричном процессе происходят ошибки.
Их называют ошибки спаривания. Эти
ошибки происходят с частотой – одна
ошибка на 2 х 104включённых
нуклеотидов. Как мы отмечали при
репликации ошибки возникают намного
реже – одна на 1010 включений. При
ошибках спаривания в растущую нить
РНК включаются «не правильные»
основания. Это может
Терминатор

Инвертированные
последовательности


 А
А
А


РНК-полимераза
 РНК-полимераза
РНК-полимераза
Б

В
5’
Рис. 57. Терминация. Образование
«шпильки» на РНК при считывании
инвертированных повторов на матричной
нити ДНК. А – РНК-полимераза «подходит»
к области терминатора. У последнего
имеется инвертированный повтор. Б –
РНК-полимераза синтезировала РНК, в
которой имеется последовательности
комплементарные инвертированному
повтору на матричной нити ДНК. В –
нуклеотиды инвертированного повтора
на РНК сформировали связи, что привело
к образованию «шпильки». Транскрипция
прекратилась.
привести к изменению структуры кодирующего
нуклеотида. Если это касается рРНК или
тРНК то, как правило, такие «дефектные»
РНК перестают выполнять свои функции
– формирование рибосом и транспорт
аминокислот. Но это практически не
отражается на здоровье человека, т.к.
появление одной «дефектной» молекулы
РНК приходится на несколько тысяч
нормальных молекул, к тому же ошибочное
спаривание очень редкое явление. Более
неблагоприятный прогноз бывает связан
с появлением ошибки спаривания в иРНК
– молекуле несущей информацию об
аминокислотной последовательности
белка. В этом случае вероятность
реализации генетического дефекта в
фенотипический определяется характером
нуклеотида образованного при ошибке
спаривания. Хорошо если в результате
«вырожденности» третьего основания
триплета, новый кодон кодирует ту же
аминокислоту. Такая ошибка фенотипически
не проявляется. (см. раздел 3.1.). Подсчитано,
что в 67% смысл кодона не меняется при
замене нуклеотида в третьем положении
кодона. Хуже когда сформированный на
основании ошибки спаривания кодон
меняет смысл и начинает кодировать
другую аминокислоту. Это приводит к
появлению в белке новой аминокислоты,
которая может изменить функциональные
свойства белка и последний выключится
из клеточного метаболизма. Но и в этом
случае новая аминокислота может по
физико-химическим свойствам (например,
растворимости) быть сравнима с заменённой.
В этом случае фенотипические последствия
такой замены менее опасны.
Но в целом необходимо отметить, что
ошибки спаривания на уровне транскрипции
менее опасны, чем на уровне репликации.
Транскрипцию могут ингибировать
различные соединения. Так например,
очень опасный шляпочный гриб бледная
поганка содержит токсин альфа-аманитин.
Три характерных признака этого гриба
необходимо помнить – бледный цвет,
«юбочка» на ножке и булавовидное
утолщение в нижней части ножки.
Этот токсин прочно связывается с
РНК-полимеразой IIу
эукариот, которая транскрибирует иРНК.
В результате блокируется транскрипция
многих генов.
Другой токсин – актиномицин Д, является
антибиотиком и прочно связывается с
ГЦ-богатыми участками ДНК. А поскольку
такими участками обогащены рРНК и
терминаторы многих генов, то эти структуры
страдают в первую очередь.
Как мы отмечали ранее некоторые
противоопухолевые препараты, например
дауномицин и некоторые другие содержат
в своей молекуле плоскую циклическую
структуру, которая встраивается
(интеркалируется) между парами оснований.
Это ведёт к локальному изменению
структуры ДНК, в результате чего ферменты
транскрипции прекращают свою работу.
Некоторые алкилирующие вещества –
тиофосфамид и др. модифицируют основания
в ДНК путём присоединениё к ним алкильных
группировок. Если ферменты транскрипции
встречают такие основания, их работа
прекращается.
Ингибитор ДНК-топоизомеразы – новобиоцин,
вмешиваясь в работу фермента прекращая
деспирализацию ДНК, а следовательно и
синтез РНК.
Антибиотик рифампицин – ингибируют
только РНК-полимеразу бактерий,
препятствуя инициации транскрипции.
Применяют его при различных заболевания,
так как он не действуют на ядерную
РНК-полимеразу соматических клеток
человека. К сожалению, в последнее время
обнаружено действие рифампицина на
РНК-полимеразу митохондрий, где также
осуществляется транскрипция митохондральных
РНК. Это является ещё одним подтверждением
версии о том, что митохондрии являются
потомками вирусных или иных
субмикроскопических структур внедрившихся
в клетки эукариот и адаптировавшихся
к их метаболизму так, что стали его
полноправными участниками.
МЗ. Транскрипция –первый этап экспрессии
генов и заключается в синтезе РНК на
одной нити ДНК ферментом ДНК-зависимой
РНК-полимеразой (РНК-полимеразой). Состоит
из трёх этапов – инициации, элонгации
и терминации. Во время инициации
формируется инициаторный комплекс,
состоящий белка прикреплённого к
промотору, РНК-полимеразы и нескольких
факторов инициации. Одновременно
формируется транскрипционный «глазок».
Во время элонгации происходит наращивание
нити РНК в соответствии с комплементарными
нуклеотидами на матричной (антисмысловой)
цепочки ДНК. Сигналом терминации является
определённая последовательность
нуклеотидов в матричной цепочки ДНК.
При транскрипции чаще, чем при репликации
возникают ошибки при спаривании
нуклеотидов.
Соседние файлы в папке лекции по биологии
- #
- #
- #
- #
- #
- #
- #
Исправление ошибок. ДНК-полимеразы могут выявлять и исправлять ошибки, тогда как РНК-полимеразы такой способностью, по-видимому, не обладают. Поскольку ошибка даже в одном основании как при репликации, так и при транскрипции может привести к ошибке в синтезе белка, можете ли вы дать биологическое объяснение этому поразительному различию [c.925]
Очень важно отметить, что процесс репликации протекает со значительно более высокой степенью точности, чем процессы транскрипции и трансляции. Частые ошибки в репликации подвергли бы большому риску сохранность видов [c.908]
События, влияющие на транскрипцию и трансляцию. Могут происходить ошибки как на уровне транскрипции, так и на уровне трансляции. [c.42]
Спонтанные генные мутации определяются ошибками при репликации ДНК, возникающими вследствие теплового движе-иия атомов и молекул. Очевидно, что ошибки транскрипции и трансляции не наследуются. [c.283]
ВЫВОД, ЧТО, по-видимому, код действительно является триплет-ным, причем кодирование начинается от определенной точки нуклеиновой кислоты. При этом большая часть трехбуквенных комбинаций соответствует определенным аминокислотам и лишь небольшая часть триплетов относится к бессмысленным. Число триплетов равно 4-4-4 = 64, т. е. значительно больше числа аминокислот. Некоторые из них, по-видимому, кодируют одну и ту же аминокислоту, т. е. код является вырожденным. Этот вывод согласуется с обнаружением в настоящее время двух и более типов растворимых РНК, специфичных к одной и той же аминокислоте. Вырожденность генетического кода может способствовать выживанию организма. Действительно, в случае невырожденного кода ошибка при репликации ДНК или при транскрипции должна скорее приводить к появлению бессмысленного триплета, чем в случае вырожденного кода. Следовательно, при невырожденном коде ошибки чаще вызывали бы прекращение синтеза соответствующего белка или образование незаконченных белковых цепей. Напротив, в случае вырожденного кода ошибки должны чаще приводить просто к замене одной аминокислоты на другую, что, как правило, не имеет серьезных последствий. [c.376]
Спонтанные генные мутации определяются ошибками, возникающими вследствие теплового движения атомов и молекул при редупликации ДНК. Очевидно, что ошибки при транскрипции и трансляции не наследуются. [c.600]
Процесс транскрипции находится в клетке под строгим контролем, поэтому имеет место как неодинаковое транскрибирование во времени разных участков ДНК (генов), так и неодинаковая скорость, с которой гены могут транскрибироваться. В результате количество молекул иРНК в клетке, комплементарных разным генам, сильно различается. Хотя в целом механизмы синтеза ДНК и РНК сходны, процесс транскрипции не обладает той степенью точности, которая характерна для репликации ДНК. Однако поскольку иРНК не способна к самовоспроизведению, возникающие при ее синтезе ошибки в последующих клеточных генерациях не воспроизводятся и, следовательно, не могут наследоваться. [c.142]
Если же программа ошибется, выбрав слишком удаленное от старта транскрипции положение блоков, вектор Г изменяется и становится равным У(,, +х , где у — вектор, соответствующий выбранному положению структуры, ах — оптимальный из всех правильных векторов. Тем самым программа не только наказывается за ошибки, но и стимулируется к распознаванию правильных положений. [c.140]
Потребность в гц)авильно спаренном конце как раз и наделяет ДНК полимеразу способностью исправлять свои собственные ошибки. Такой фермент, очевидно, мог бы начать синтез ДНК при полном отсутствии затравки только утратив способность различать спаренный и неспаренный концы В то же время РНК-полимеразы, участвующие в транскрипции генов (см. разд. 5.1.1) судя по всему не нуждаются в само коррекции, потому что ошибки транскрипции не передаются следующему поколению и случайно возникшие дефектные молекулы особой роли не играют. РНК-полимеразы могут начинать синтез новых полинуклеотидных цепей в отсутствие затравки, причем ошибки встречаются с частотой 10 как при синтезе РНК, так и при трансляции, т е. при переводе нуклеотидных последовательностей мРНК в аминокислотные последовательности белков. [c.290]
Процесс синтеза комплементарной ДНК по РНК-мап-рице ферментом обратная транскриптаза . Процесс, склонный к ошибкам, потому что нет редактирования вновь синтезируемой ДНК и, следовательно, могут накапливаться мутации. Обратная транскрипция противоположна нормальному движению генетической информации от ДНК к РНК. См. Центральная догма [c.16]
Идущее различными путями умножение копий гена в хромосоме представляет материал для усиления функций изменения генов. Первое понятно сразу. А вот второе… тут есть два соображения. Во-первых, когда экземпляров гена несколько, то часть из них может меняться, а жизнеспособность организма при этом практически сохранится — функцию выполнят другие. Во-вторых, сам процесс удвоения может вносить ошибки. Так, например, если повтор получен в результате обратной транскрипции РНК ДНК, то вероятность ошибки примерно в миллион раз больше, чем при обычном удвоении ДНК ДНК [34. [c.147]
На специфическом подавлении отдельных стадий биосинтеза белка основано действие ряда антибиотиков (разд. 2.3.5). Так, актиномицин интер-коляцией и рифамицин селективным подавлением РНК-полимеразы нарушают процесс транскрипции. Хлорамфеникол нарушает трансляцию, блокируя реакцию переноса пептидила в рибосоме. Стрептомицин ассоциирует с 30 8-субъединицей рибосомы и ведет к ошибкам в переносе, а очень похожий на аминоацильный конец тРНК пуромицин вызывает преждевременный обрыв синтезируемой цепи. [c.398]
К счастью, с РНК-полимеразой такой ошибки не произошло. Она действительно оказалась тем самым ферментом, который ведает в клетке транскрипцией. Однако открытие этого фермента отвечало отнюдь не на все вопросы, связанные с синтезом мРНК. В самом деле, РНКовая копия снимается каждый раз не со всей ДНК, а с ее небольшого участка, содержащего один или несколько генов. Что же происходит с другими генами Если они молчат, то почему Может быть, есть не одна, а много РНК-полимераз, которым положено читать разные гены Или, может быть, существуют еще другие белки (назовем их репрессорами), которые не подпускают РНК-полимеразу к молчащим генам, не дают их считывать Какое объяснение предпочесть [c.50]
Идентификация антикодона в тРНК проведена весьма убедительными экспериментами. Получены мутанты, содержаш,ие мутационную ошибку в самом антикодоне определенной тРНК. Затем путем полного химич. анализа цепи тРНК показано, какой именно нуклеотид оказался замененным. Здесь и находился антикодон. Связь кодон — антикодон осуществляется тремя нуклеотидами, а не длинной цепочкой. Поэтому возможность ошибок, или уровень шумов , в процессе трансляции (так называют синтез белка) выше, чем при редупликации ДНК или транскрипции РНК. Вероятность ошибки при наборе белковой цепи достигает 10 (вместо 10 при матричном синтезе дезоксирибонуклеиновой кислоты). [c.196]
Синтез вирусной РНК начинается всегда на З -конце РНК-матрицы (т. е. с 5 -конца новой молекулы РНК) и вдет до тех пор, пока не будет достигнут 5 -конец матрицы. Никаких механизмов, которые корректировали бы синтез вирусной РНК, нет, и частота ошибок здесь примерно та же, что и при транскрипции ДНК (в среднем одна ошибка на Ю нуклеотвдов). Однако отсутствие корректирующих механизмов серьезным образом не сказывается на репликации из-за небольших размеров РНК-хромосомы вируса Геномы всех РНК-вирусов невелики в сравнении с геномами крупных ДНК-содержащих вирусов, и это является прямым следствием того, что у них механизм репликации более примитивен. [c.317]
Феномен старения и смерти живого организма предопределен биологически и обеспечивает общее эволюционное развитие живой природы. В соответствии с законом естественного отбора в ходе эволюции не только расширяются метаболические (трофические) связи живых систем с окружающей средой, но и увеличивается информационное содержание этих связей. Информационное обеспечение в свою очередь повышает надежность функционирования каждого живого организма и его соответствие окружающей среде на данном этапе эволюции (Шмальгаузен, 1961). Таким образом, изменчивость, вариабельность, накопление новых биологических свойств позволяют живым системам приспосабливаться к изменениям окружающей среды, однако трансляция этих изменений в последующие поколения строго Офаничена консерватизмом генетического аппарата. Старение организма определяется понижением информационной надежности генетического аппарата живых клеток в результате накопления ошибок при репликации ДНК и транскрипции генетической информации, что в свою очередь приводит к ошибкам при синтезе и процессинге полипептидов и белков (Бриллюэн, 1966 Сьяксте, Будылин, 1992). Стратегия биологической эволюции заключается в том, что организм, в котором накопление молекулярных дефектов генетического аппарата достигло критического уровня, изымается из популяции. Иными словами, наряду с этапами зачатия, роста и развития организма старение является эндогенно обусловленным, т. е. естественным, терминальным этапом. Оно начинается периодом стабилизации жизненных функций, угасанием репродуктивного потенциала, постепенным замедлением метаболизма и завершается периодом снижения активности и отмиранием отдельных клеточных систем и тканей. Отказ одних систем организма, как пра- [c.159]
Открытие ферментативных функций РНК и ее способности к саморепликации завершило 30—40-летний период попыток рационально объяснить первые шаги молекулярной эволюции жизни. Тот факт, что одноцепочечные молекулы РНК способны сами себя реплицировать и изменять, представлялся с функциональной точки зрения существенным. Сейчас известно, что все матричные процессы копирования, затрагивающие РНК (транскрипция, репликация и обратная транскрипция — рис. 2.4 А, В), склонны к ошибкам. [c.60]
Мы описали протекающий в В-лимфоцитах процесс обратной связи V(D)J-reHOB, основанный на склонной к ошибкам обратной транскрипции. Он составляет основу определяемого антигеном мутирования генов антител. Все экспериментальные данные согласуются с этой теорией. Это приводит нас ко второй неола-маркистской концепции направленной обратной связи генов. [c.147]
Молекулярная биология клетки — Том 1 — Албертс Б., Брей Д., Льюис Дж., Рэфф М., Робертс К., Уотсон Дж. 1994
Молекулярная организация клеток
Основные генетические механизмы
Механизмы репликации ДНК
Живые организмы должны не только поддерживать целостность нуклеотидных последовательностей ДНК путем ее репарации, но еще и очень точно воспроизводить свою ДНК перед каждым клеточным делением. При репликации ДНК скорость полимеризации колеблется в пределах от 500 нуклеотидов в 1 с у бактерий приблизительно до 50 нуклеотидов у млекопитающих. Ясно, что ферменты, катализирующие процесс репликации, должны работать и точно, и быстро. Быстрота и точность достигаются с помощью особого мультиферментного комплекса, направляющего процесс репликации. Этот комплекс, состоящий из нескольких различных белков, представляет собой сложный и совершенный «аппарат репликации».
5.3.1. Репликация ДНК, как и ее репарация, основана на комплементарном спаривании оснований [25]
Матричная активность ДНК проявляется в том, что ее нуклеотидная последовательность копируется (целиком или частично) путем комплементарного спаривания оснований (А с Т или G с С) в виде комплементарной последовательности нуклеотидов ДНК или РНК. Этот процесс предполагает узнавание каждого нуклеотида в ДНК свободным (неполимеризованным) комплементарным нуклеотидом и обязательное разделение (хотя бы на время) двух цепей ДНК, с тем чтобы в каждом основании группы, играющие роль доноров и акцепторов при образовании водородных связей, оказались доступными для комплементарного спаривания. Таким образом поступающие одиночные нуклеотиды выстраиваются в определенном порядке вдоль матричной цепи ДНК для ферментативной полимеризации, продуктом которой является новая полинуклеотидная цепь. В 1957 г. был открыт первый фермент, катализирующий процесс полимеризации нуклеотидов; он был назван ДНК-полимеразой. Было показано, что субстратами ДНК-полимеразы служат дезоксирибонуклеозидтрифосфаты, полимеризующиеся на одно-цепочечной ДНК-матрице (двухступенчатый механизм этой полимеризации представлен на рис. 5-34 в связи с обсуждением процесса репарации ДНК). Позже была выделена и РНК-полимераза, для которой субстратами служат рибонуклеозидтрифосфаты.
Во время репликации ДНК каждая из двух ее старых цепей служит матрицей для образования новой цепи. Поэтому чрезвычайно длинная нуклеотидная последовательность клеточной ДНК реплицируется, как это принято называть, «полуконсервативно» и каждая из двух дочерних клеток получает при клеточном делении новую двойную спираль ДНК, состоящую из одной старой и одной новой цепи (см. рис. 3-11).
5-23
5.3.2. Репликационная вилка асимметрична [26]
Исследования, проведенные в начале 1960-х годов на реплицирующихся хромосомах, в которые в качестве импульсной метки вводили радиоактивный предшественник ДНК 3Н-тимидин, выявили особую четко ограниченную область репликации, перемещающуюся вдоль родительской спирали ДНК. Эта активная область из-за своей Y-образной формы была названа репликационной вилкой. Именно в ней с помощью мультиферментного комплекса, содержащего ДНК-полимеразу, синтезируются дочерние молекулы ДНК.
В то время казалось вполне вероятным, что простейший механизм репликации ДНК заключается в непрерывном росте обеих новых цепей нуклеотид за нуклеотидом по мере перемещения репликационной вилки от одного конца молекулы ДНК к другому. Однако, поскольку две цепи в спирали ДНК антипараллельны, одна из дочерних цепей должна расти в направлении 5′ → 3′, а другая — в направлении 3′ → 5′. В таком случае репликационной вилке потребовалось бы две разные ДНК-полимеразы. Одна из них наращивала бы цепь в направлении 5′ → 3′ (рис. 5-34); при этом каждый поступающий мономер (дезоксирибонуклеозидтрифосфат) приносит с собой необходимую для его присоединения к цепи энергию (ее носителем является трифосфатная группа). Другая ДНК-полимераза, перемещающаяся в направлении 3′ → 5′, должна катализировать «рост с головы»; в этом случае энергию, необходимую для присоединения каждого очередного нуклеотида, должен нести конец растущей цепи ДНК. В действительности такой (3′ → 5′) ДНК-полимеразы не существует (рис. 5-38), хотя биохимикам известны некоторые другие процессы полимеризации, протекающие по типу «роста с головы» (см. рис. 2-34), Каким же образом происходит рост цепи в направлении 3′ → 5′? Возможный ответ на этот вопрос подсказали в конце 1960-х годов эксперименты с радиоактивно меченными предшественниками ДНК. Если растущие клетки получают всего на несколько секунд высокорадиоактивный 3Н-тимидин, то метка включается лишь в ДНК, синтезированную в самый последний момент, т. е. в ту ее часть, которая следует непосредственно за репликационной вилкой. Этим методом избирательного введения метки было выявлено, что при репликации бактериальной ДНК в области репликационной вилки образуются и какое-то время существуют фрагменты, насчитывающие от 1000 до 2000 нуклеотидов (впоследствии за ними закрепилось название «фрагменты Оказаки»; у эукариот они гораздо короче: от 100 до 200 нуклеотидов). Несколько позже было показано, что синтез этих фрагментов ДНК идет только в направлении 5′ → 3′; синтезированные фрагменты соединяются затем в длинные цепи ДНК под действием того же фермента, который сшивает разрывы в спирали ДНК во время ее репарации, т.е. под действием ДНК-лигазы (см. рис. 5-35).
Рис. 5-38. На первый взгляд простейшим механизмом репликации ДНК представляется механизм, изображенный на этой (неверной!) схеме. Обе дочерние цепи должны были бы при этом расти непрерывно за счет присоединения нуклеотидов соответственно в 5′ → 3′ — направлении (на рисунке — внизу) и 3′ → 5′ — направлении (на рисунке — вверху). Однако фермента, который бы катализировал присоединение нуклеотидов в направлении 3′ → 5′, не существует.
Рис. 5-39. Строение репликационной вилки. Обе дочерние цепи строятся в направлении 5′ → 3′. Для этого отстающая цепь ДНК должна синтезироваться в виде ряда коротких фрагментов (фрагменты Оказаки).
Репликационная вилка асимметрична (рис. 5-39). Из двух синтезируемых дочерних цепей ДНК одна строится непрерывной, а другая прерывистой. Первую называют ведущей (или лидирующей), а вторую — отстающей. Наращивание второй цепи отстает, потому что образование каждого фрагмента Оказаки оказывается возможным лишь после того, как продвижение ведущей цепи откроет соответствующий участок матрицы. Хотя в целом вся эта цепь строится в направлении 3′ → 5′, каждый из ее фрагментов синтезируется в направлении 5′ → 3′. Благодаря тому что ДНК на отстающей части вилки строится при помощи механизма, работающего прерывисто по типу «шитья назад иголкой» (backstitching), в репликационной вилке не требуется никакого другого фермента, кроме (5′ → 3′)-ДНК-полимеразы.
5-24
5.3.3. Высокая точность репликации ДНК предполагает наличие механизма, осуществляющего коррекцию [27]
Точность копирования при репликации ДНК столь велика, что в среднем на каждые 1-109 комплементарных пар, образующихся в процессе воспроизведения генома млекопитающих, насчитывающего 3-104 пар оснований (см. разд. 9.1.3), приходится приблизительно одна ошибка. Точность эта значительно превосходит ту, какую следует ожидать, учитывая, что во время репликации образуются не только обычные комплементарные пары оснований. В нормальной ДНК возникают на короткое время с частотой 10-4-10-5 редкие таутомерные формы всех четырех ее оснований. Эти формы образуют неправильные пары. Так, редкая таутомерная форма С спаривается с А вместо G, в результате чего возникает мутация (рис. 5-40). Таким образом высокая точность репликации ДНК определяется наличием механизмов, осуществляющих коррекцию, т. е. устраняющих подобные ошибки.
Рис. 5-40. Пример возникновения при репликации ДНК неправильной пары оснований: находясь в термодинамически невыгодной таутомерной форме, цитозин легко образует водородные связи с аденином.
Рис. 5-41. Схема, поясняющая, как протекает процесс коррекции (устранение ошибок) при синтезе ДНК, катализируемом ДНК-полимеразами у бактерий. Предполагается, что аналогичный механизм коррекции действует и в эукариотических клетках.
Один из важных механизмов коррекции зависит от особых свойств ДНК-полимеразы. В отличие от РНК-полимераз ДНК-полимеразы не могут начать синтез новой полинуклеотидной цепи, просто связав друг с другом 3′-ОН- конец какой-либо полинуклеотидной цепи, которая должна быть спарена с матричной цепью ДНК; ДНК-полимеразы способны только добавлять новые нуклеотиды к уже имеющемуся 3′-ОН-концу полинуклеотидной цепи (см. рис. 534). Эту предобразованную цепь, к которой добавляются нуклеотиды, называют затравкой или праймером. Молекулы ДНК с затравкой, у которой 3′-ОН-конец не спарен, не могут служить матрицами. Бактериальные ДНК-полимеразы способны, однако, с ними работать. Вступив в контакт с такими молекулами ДНК, они используют присущую им (3′ → 5′)-экзонуклеазную активность и отщепляют (путем гидролиза) любые неспаренные нуклеотиды на затравочном конце. Отщепляется ровно столько нуклеотидов, сколько требуется для того, чтобы у затравки появился спаренный конец и образовалась активная матрица. Действуя таким образом, ДНК-полимераза выступает в роли «самокорректирующего» фермента: она устраняет свои собственные ошибки, возникающие в процессе полимеризации. Рис. 5-41 поясняет, как этот тип коррекции может использоваться для удаления неправильных пар С—А, образуемых редкой таутомерной формой цитозина.
Потребность в правильно спаренном конце как раз и наделяет ДНК- полимеразу способностью исправлять свои собственные ошибки. Такой фермент, очевидно, мог бы начать синтез ДНК при полном отсутствии затравки, только утратив способность различать спаренный и неспаренный концы. В то же время РНК-полимеразы, участвующие в транскрипции генов (см. разд. 5.1.1), судя по всему не нуждаются в самокоррекции, потому что ошибки транскрипции не передаются следующему поколению и случайно возникшие дефектные молекулы особой роли не играют. РНК-полимеразы могут начинать синтез новых полинуклеотидных цепей в отсутствие затравки, причем ошибки встречаются с частотой 10-4 как при синтезе РНК, так и при трансляции, т.е. при переводе нуклеотидных последовательностей мРНК в аминокислотные последовательности белков.
5-22
5.3.4. Репликация ДНК в направлении 5’ → 3’ обеспечивает эффективную коррекцию
Весьма вероятно, что однонаправленность репликации ДНК (5′ → 3′) объясняется высокими требованиями к точности процесса. Если бы существовала ДНК-полимераза, присоединяющая дезоксирибонуклеозидтрифосфаты к синтезируемой полинуклеотидной цепи таким образом, что эта цепь росла в направлении 3′ → 5′, то активирующую трифосфатную группировку нес бы растущий 5′-конец цепи, а не поступающий мононуклеотид. В этом случае ошибки полимеризации не могли бы устраняться простым гидролизом, потому что появление свободного 5′-конца немедленно обрывало бы синтез ДНК. Ясно, что основание, только что неправильно спарившееся на 3′-конце, устранить гораздо легче, чем такое же основание, присоединившееся к 5′-концу цепи ДНК. Поэтому, хотя механизм репликации ДНК, изображенный на рис. 5-39, кажется на первый взгляд значительно более сложным и громоздким, чем неверный гипотетический механизм, представленный на рис. 5-38, этот реально функционирующий механизм способен обеспечить гораздо большую точность именно в силу того, что синтез ДНК идет здесь только в направлении 5′ → 3′.
5-25
5.3.5. Для синтеза коротких затравочных молекул на матрице отстающей цепи требуется особый фермент [28]
С того момента, как возникла репликационная вилка, для ДНК-полимеразы, синтезирующей ведущую цепь, всегда есть спаренный 3′-конец, необходимый ей для того, чтобы начать синтез новой цепи. Иначе обстоит дело с ДНК-полимеразой, ответственной за синтез отстающей цепи. Ей требуется всего каких-нибудь 4 с для того, чтобы синтезировать один короткий фрагмент ДНК, после чего она должна переключиться на синтез совсем другого фермента на новом участке матричной цепи, расположенной на некотором расстоянии от первого (см. рис. 5-39). Для этого ей всякий раз нужна затравка со спаренным 3′-концом, а следовательно, нужен и механизм, способный производить такие затравки. В этот механизм входит фермент, называемый ДНК-праймазой. Она синтезирует из рибонуклеозидтрифосфатов короткие РНК-затравки (праймеры), состоящие у эукариот примерно из 10 нуклеотидов (рис. 5-42). Эти затравки синтезируются с определенными интервалами на матрице для отстающей цепи; здесь их наращивает ДНК-полимераза, начиная, таким образом, всякий раз новый фрагмент Оказаки. Молекула ДНК-полимеразы продолжает это наращивание до тех пор, пока она не достигнет РНК-затравки, присоединенной к 5′-концу предыдущего фрагмента ДНК. Чтобы обеспечить образование непрерывной цепи ДНК из многих таких фрагментов, в действие вступает особая система репарации ДНК, быстро удаляющая РНК-затравку и заменяющая ее на ДНК. Завершает процесс ДНК-лигаза, соединяющая 3′-конец нового фрагмента ДНК с 5′-концом предыдущего фрагмента (рис. 5-43).
Почему предпочтение отдается удаляемой РНК-затравке, а не ДНК-затравке, которую не требовалось бы удалять? Выше мы отмечали, что самокорректирующая полимераза не способна начинать синтез полинуклеотидных цепей de novo; это предполагает и обратное утверждение: тот фермент, который начинает синтез цепей de novo, к эффективной самокоррекции не способен. Значит, любой фермент, катализирующий инициацию синтеза фрагментов Оказаки, неизбежно создал бы не слишком точную копию (не менее 1 ошибки на 105). Это означало бы колоссальное увеличение частоты мутаций даже при том, что количество таких копий, сохранившееся в конечном продукте, составляло бы не более 5% всего генома (например, 10 нуклеотидов во фрагменте, состоящем из 200 нуклеотидов). Естественно думать поэтому, что выдвижение РНК, а не ДНК на роль затравки обеспечивало важное преимущество, поскольку рибонуклеотиды автоматически метят такие последовательности, как «плохие копии», которые должны быть удалены.
Рис. 5-42. Схема реакции, катализируемой праймазой — ферментом, синтезирующим короткие РНК-затравки в отстающей цепи ДНК. В отличие от ДНК-полимеразы этот фермент способен начинать синтез новой полинуклеотидной цепи с соединения двух нуклеозидтрифосфатов. Образовав короткий полинуклеотид, праймаза прекращает работу. Теперь к свободному 3′-концу может добавлять нуклеотиды ДНК-полимераза.
5-26
5-33
5.3.6. Особые белки способствуют расплетанию двойной спирали ДНК перед репликационной вилкой [29]
Двойная спираль ДНК должна расплетаться по ходу продвижения репликационной вилки, для того чтобы поступающие дезоксирибонуклеозидтрифосфаты могли спариваться с родительской матричной цепью. Однако в обычных условиях двойная спираль ДНК весьма стабильна; спаренные основания соединены столь прочно, что для разделения двух цепей ДНК в пробирке требуются температуры, приближающиеся к точке кипения воды (90°С). По этой причине большинство ДНК-полимераз может копировать лишь ту молекулу ДНК, у которой матричная цепь уже отделилась от другой цепи. Для того чтобы двойная спираль ДНК раскрылась и соответствующая матричная цепь стала доступной для ДНК-полимеразы, необходимы особые белки. Они бывают двух типов.
ДНК-геликазы были впервые выделены как белки, которые, присоединяясь к одиночной цепи ДНК, катализируют гидролиз АТР. Как уже отмечалось в гл. 3, гидролиз АТР может циклическим образом изменять форму молекулы белка, вследствие чего белок будет производить механическую работу (см. разд. 3.4.11). Именно этот принцип лежит в основе быстрого перемещения ДНК-геликаз по одиночной цепи ДНК. Встречая на своем пути участок двойной спирали, эти ферменты продолжают двигаться вдоль своей цепи и тем самым расплетают двойную спираль (рис. 5-44). Расплетание ДНК-спирали в области репликационной вилки, вероятно, осуществляется двумя совместно действующими ДНК- геликазами, одна из которых перемещается по ведущей цепи, а другая — по отстающей. Ясно, что две эти геликазы должны двигаться вдоль одиночных цепей ДНК в противоположных направлениях, т. е. это должны быть разные ферменты. Действительно, оба указанных типа ДНК- геликаз удалось обнаружить. При этом исследования на бактериях показали, что главную роль играет ДНК-геликаза отстающей цепи. Причины этого мы обсудим ниже.
Белки, дестабилизирующие спираль (их называют также белками, связывающими одноцепочечную ДНК или SSB-белками), связываются с одиночными цепями ДНК, не закрывая оснований, т. е. оставляя их доступными для спаривания. Сами они не способны расплетать длинные молекулы ДНК, но, присоединяясь к одиночным цепям ДНК, они тем самым способствуют любому процессу расплетания спирали; они, например, помогают ДНК-геликазе расплетать двойную спираль в репликационной вилке. На матрице отстающей цепи SSB-белки кооперативным образом связываются с одноцепочечными участками ДНК и предотвращают здесь образование «шпилек», небольших двухспиральных структур, которые могли бы помешать синтезу ДНК, осуществляемому ДНК-полимеразой (рис. 5-45).
Рис. 5-43. Отдельные этапы синтеза каждого из фрагментов отстающей цепи ДНК. У эукариот РНК-затравки синтезируются в отстающей цепи с интервалами приблизительно в 200 нуклеотидов и каждая из них состоит из 10 нуклеотидов.
5.3.7. Белки в репликационной вилке действуют кооперативно, образуя «репликационную машину» [30]
До сих пор мы говорили о репликации ДНК так, как если бы она осуществлялась смесью репликационных белков, действующих независимо друг от друга. Между тем в действительности большая часть этих белков объединена в крупный мультиферментный комплекс, быстро движущийся вдоль ДНК. Этот комплекс — нечто вроде крошечной «швейной машины»: «деталями» его служат отдельные белки, а источником энергии — реакция гидролиза нуклеозидтрифосфата. Комплекс изучен достаточно хорошо только у бактерий Е. coli и у некоторых вирусов, но есть все основания считать, что очень похожий механизм действует и у эукариот (см. разд. 9.3.3).
Схема на рис. 5-46, где подробно изображена репликационная вилка, позволяет судить о том, как работают отдельные части такой «репликационной машины». В области вилки действуют две идентичные ДНК-полимеразы — на ведущей и на отстающей цепи. Спираль ДНК расплетается в результате совместного действия ДНК-полимеразы, работающей на ведущей цепи, и ДНК-геликазы, движущейся вдоль отстающей цепи; этому процессу способствуют кооперативно связывающиеся молекулы дестабилизирующегося белка. В то время как на ведущей цепи ДНК- полимераза работает непрерывно, на отстающей цепи фермент через определенные интервалы прерывает и вновь возобновляет свою работу, используя для полимеризации короткие РНК-затравки, синтезируемые ДНК-праймазой.
Эффективность репликации сильно возрастает вследствие тесного объединения всех этих белковых компонентов. Молекула праймазы непосредственно сцеплена с ДНК-геликазой, образуя вместе с нею на отстающей цепи структуру, называемую праймосомой, которая движется с репликационной вилкой и по ходу своего движения синтезирует РНК-затравки. Молекула ДНК-полимеразы, работающая на отстающей цепи, также движется совместно с остальными белками, синтезируя ряд новых фрагментов Оказаки; ради этого, как полагают, цепь ДНК, которая служит для нее матрицей, складывается сама на себя, как это показано на рис. 5-47. Репликационные вилки оказываются, таким образом, объединены в одну крупную структуру (с общей массой > 106 дальтон), быстро перемещающуюся вдоль ДНК и обеспечивающую возможность координированного и эффективного синтеза ДНК на обет ветвях вилки.
Рис. 5-44. Действие ДНК-геликаз. Небольшой фрагмент ДНК присоединен путем отжига к длинной одноцепочечной ДНК, так что образовался короткий участок двойной спирали. Эта спираль расплетается по мере того, как геликаза движется вдоль одиночной цепи ДНК, катализируя реакцию, для которой требуется наряду с ферментом и АТР. Источником энергии для движения геликазы служит гидролиз АТР (см. рис. 3-63).
Рис. 5-45. Влияние дестабилизирующих белков на структуру одно-цепочечной ДНК. Поскольку каждая белковая молекула предпочитает связываться с другой, уже связавшейся ранее молекулой (так называемое кооперативное связывание), эти белки образуют длинные кластеры, выпрямляющие матричные пени ДНК и облегчающие процесс полимеризации. Структуры в форме «шпильки», возникающие в свободной одноцепочечной ДНК, образуются путем случайного спаривания оснований в коротких участках, содержащих взаимно комплементарные последовательности нуклеотидов, они напоминают короткие спирали, возникающие во всех молекулах РНК.
Рис. 5-46. Главные типы белков, действующих в области репликационной вилки (схема показывает их локализацию на ДНК). Комплекс ДНК-праймазы и ДНК-геликазы на отстающей цепи ДНК известен под названием праймосомы.
Позади «репликационной машины» по ходу ее движения остается на отстающей цепи ряд несшитых фрагментов Оказаки, все еще содержащих на своем 5′-конце РНК-затравки, необходимые для инициации на синтеза. Эти РНК-затравки должны быть удалены, а фрагменты сшиты при помощи репарирующих ферментов, работающих позади репликационной вилки (см. рис. 5-43).
Рис. 5-47. Схема, иллюстрирующая современные представления о расположении репликационных белков в движущейся репликационной вилке. Вместо двумерной структуры, изображенной на рис. 5-46, здесь показано, как ДНК на отстающей цепи складывается, в результате чего возникает комплекс из двух ДНК-полимераз — ведущей и отстающей цепи. Кроме того, благодаря складыванию 3′-конец каждого завершенного фрагмента Оказаки оказывается рядом со стартовым участком следующего такого фрагмента (ср. с рис. 5-46). Находясь в тесном контакте с остальными репликационными белками, молекула ДНК-полимеразы отстающей цепи может непрерывно работать на одной и той же репликационной вилке, отделяясь от готового фрагмента ДНК, она переходит к ближайшей новой РНК-затравке, чтобы начать синтез следующего фрагмента. Обратите внимание, что на этой схеме одна из дочерних спиралей ДНК направлена вправо и вниз, а вторая влево и вверх.
5.3.8. Ошибки при репликации ДНК в бактериальных клетках устраняются особой корректирующей системой, распознающей неправильное спаривание оснований
У таких бактерий, как Е. coli, деление происходит каждые 30 мин, поэтому у них сравнительно легко выявить в большой популяции клеток редкие экземпляры с измененными признаками. Выделен, например, класс мутантов, характеризующихся резким повышением частоты спонтанных мутаций, что связано с присутствием в его клетках специфических генов-мутаторов. Известен ген-мутатор, кодирующий дефектную форму 3′ → 5′-корректирующей экзонуклеазы, представляющей собой субъединицу ДНК-полимеразы (см. разд. 5.3.3). Если дефект затрагивает этот белок, то ДНК-полимераза утрачивает способность эффективно осуществлять коррекцию и в ДНК накапливается много ошибок, которые при нормальной репликации были бы устранены.
Изучение тех мутантов Е. coli, у которых имеются гены-мутаторы, выявило еще одну систему, в норме устраняющую ошибки репликации, не улавливаемые корректирующей экзонуклеазой. Эта система коррекции неправильного спаривания (mismatch proofreading sistem), называемая также системой исправления ошибок спаривания (mismatch repair system), отличается от ранее рассмотренных систем репарации ДНК тем, что она не зависит от присутствия в ДНК аномальных нуклеотидов, которые должны быть распознаны и удалены («вырезаны»). Она выявляет деформации на внешней стороне спирали, вызванные плохой пригонкой обычных, но некомплементарных оснований. Если бы эта корректирующая система просто распознавала ошибки спаривания в реплицировавшейся ДНК и удаляла без выбора один из двух неправильно спарившихся нуклеотидов, то в половине случаев она бы сама совершала ошибку, «исправляя» не новосинтезированную, а матричную цепь, так что в среднем частота ошибок оставалась бы прежней. Для эффективной коррекции система должна уметь различать неправильно спаривающиеся нуклеотиды и избирательно удалять такие нуклеотиды только из новой цепи (т.е. устранять именно ошибки репликации).
В клетках Е. coli процесс распознавания связан с метилированием определенных остатков аденина в ДНК. Метальные группы присоединяются ко всем остаткам А в последовательности GATC, но лишь спустя некоторое время после того, как А включится в новосинтезированную цепь ДНК. Новые цепи отличаются от старых тем, что только в них сразу же за репликационной вилкой могут находиться еще не метилированные последовательности GATC. Коррекция неправильного спаривания осуществляется крупным мультиферментным комплексом, сканирующим каждую из двух цепей двойной спирали ДНК. Этот комплекс удаляет только неправильно присоединенные нуклеотиды, но делает это лишь после того, как на той же цепи обнаружится и неметилированная последовательность GATC. Поэтому нуклеотиды удаляются только из новой цепи, т. е. устраняются ошибки репликации (рис. 5-48).
Рис. 5-48. Схема эксперимента, иллюстрирующего работу системы коррекции неправильного спаривания, устраняющей у бактерий ошибки репликации ДНК. Особый белковый комплекс удаляет неспаренные нуклеотиды из вновь синтезируемой цепи ДНК позади репликационной вилки, этот репарирующий комплекс узнает новую цепь ДНК по обнаруживаемым в ней неметилированным последовательностям GATC. На схеме представлены три молекулы ДНК с одной и той же «неправильной» парой нуклеотидов, но при этом в одной молекуле (А) метилированные последовательности GATC встречаются в обеих цепях, в другой молекуле (Б) таких метилированных последовательностей нет совсем, а в третьей (В) они присутствуют только в одной из цепей. Если воздействовать на эти молекулы ДНК клеточным экстрактом, содержащим корректирующий комплекс, то мы получим представленный здесь результат. Молекула ДНК в правой части рисунка воспроизводит картину, обнаруживаемую непосредственно за репликационной вилкой: нижняя цепь соответствует новой цепи, где метилирование еще не произошло.
В эукариотических клетках не удалось пока выявить ни одного из этих двух механизмов коррекции, обнаруженных у бактерий. Однако степень точности репликации у млекопитающих и у Е. coli приблизительно одинакова, и потому можно думать, что оба описанных типа коррекции существуют и у эукариот. Следует, впрочем, отметить, что в ДНК млекопитающих нет метилированных остатков А, поэтому механизм, который используется системой репарации ошибок спаривания для узнавания новосинтезированной цепи, должен быть в данном случае иным.
5-27
5.3.9. Репликационные вилки возникают в точках начала репликации [32]
И у бактерий, и у млекопитающих образование репликационных вилок начинается с возникновения особой структуры, называемой репликационным глазком (replication bubble). Это небольшой участок, в котором две цепи родительской спирали ДНК отделились одна от другой и были использованы в качестве матриц для синтеза ДНК (рис. 5-49). Для бактерий и некоторых вирусов, размножающихся в эукариотических клетках, удалось показать, что репликационный глазок образуется в тех местах молекулы ДНК, где находятся специфические нуклеотидные последовательности, получившие название точек начала репликации. Эти последовательности состоят приблизительно из 300 нуклеотидов. Предполагают, что аналогичные точки начала репликации существуют и в эукариотических хромосомах, однако надежных доказательств этого пока нет (см. разд. 9.3.2).
Процесс возникновения репликационных вилок удалось в некоторых случаях воспроизвести in vitro. Эти опыты показали, что у бактерий и бактериофагов инициация репликационных вилок начинается так, как это представлено на рис. 5-50. Множество копий инициаторного белка связываются с особыми участками в точке начала репликации, образуя крупный белковый комплекс. Этот комплекс присоединяет затем ДНК-геликазу и помещает ее на свободную одиночную цепь ДНК в прилегающем участке спирали. Присоединяется также ДНК-праймаза, т. е. образуется праймосома, которая, двигаясь от точки начала репликации, синтезирует РНК-затравку, что дает возможность начать синтез первой цепи ДНК. Остальные белки быстро объединяются после этого в два репликационных белковых комплекса, которые теперь движутся от точки начала репликации в противоположных направлениях (см. рис. 5-49); они продолжают синтезировать ДНК до тех пор, пока обе вилки не пройдут путь по матрице до самого конца.
Некоторые дополнительные данные, касающиеся инициации репликационных вилок в хромосомах эукариот, мы обсудим в гл. 9, там, где речь пойдет о клеточном ядре.
Рис. 5-49. Гипотетический механизм образования репликационных вилок в точках начала репликации (см. также рис. 5-50).
Рис. 5-50. Упрощенная схема, иллюстрирующая начальные этапы образования репликационных вилок в точках начала репликации у Е. coli и бактериофага X. Для обнаружения данного механизма потребовались опыты in vitro с использованием смеси высокоочищенных белков.
Последующие этапы приводят (пока не ясным путем) к инициации еще трех цепей ДНК (рис. 5-49). У Е. coli в репликации ДНК роль инициаторного белка играет белок dnaA; а праймосома состоит из белков dnaB (ДНК-геликаза) и dnaG (ДНК-праймаза).
5.3.10. ДНК-топоизомеразы предотвращают спутывание ДНК во время репликации [33]
Изображая спираль ДНК так, как мы это делали до сих пор, т.е. неправильно, в виде плоской «лестницы», мы игнорировали «проблему закручивания» (winding problem). Между тем на каждые 10 пар оснований, образующихся в репликационной вилке, родительская двойная спираль должна совершить один полный оборот вокруг своей оси. Следовательно, для того чтобы репликационная вилка могла продвигаться вперед, вся хромосома впереди нее должна быстро вращаться (рис. 5-51), что для длинных хромосом потребовало бы большой затраты энергии. При репликации ДНК эта проблема решается иначе: путем образования в спирали своего рода «шарнира», особого класса белков, называемых ДНК- топоизомеразами.
ДНК-топоизомераза представляет собой нечто вроде «обратимой нуклеазы». Сначала она разрывает цепь ДНК, а затем ковалентно присоединяется к разорванному концу. Ковалентная связь белок — ДНК обладает довольно значительной энергией, потому что в ней сохраняется энергия разорванной фосфодиэфирной связи. Вследствие этого реакция, приводящая к разрыву цепи, обратима и не требует дополнительной затраты энергии. В этом отношении данный механизм существенно отличается от механизма действия ДНК-лигазы, о котором мы говорили выше (см. рис. 5-35).
Существуют различные типы ДНК-топоизомераз. Топоизомераза типа I разрывает только одну из двух цепей двойной спирали ДНК, что дает возможность двум участкам ДНК по обе стороны от разрыва свободно вращаться относительно друг друга вокруг фосфодиэфирной связи, находящейся напротив разрыва, которая в этом случае выполняет роль упомянутого выше «шарнира» (рис. 5-52). Всякое напряжение в спирали ДНК заставляет ее вращаться в таком направлении, чтобы ослабить это напряжение. Поэтому вращение во время репликации ДНК происходит лишь на коротком отрезке спирали — в той части, которая находится непосредственно перед репликационной вилкой. Аналогичная проблема, возникшая в процессе транскрипции ДНК, решается таким же путем.
Рис. 5-51. «Проблема кручения», возникающая при репликации ДНК. Для того чтобы репликационная вилка (у бактерий) могла продвигаться вперед со скоростью 500 нуклеотидов в 1 с, родительская спираль ДНК перед вилкой должна вращаться со скоростью 50 об/с.
Рис. 5-52. Обратимая реакция, приводящая к появлению разрыва в одной из цепей ДНК. Реакция у эукариот катализируется ДНК-топоизомеразой типа I Ферменты этой группы образуют временную ковалентную связь с ДНК.
Рис. 5-53. Пример реакции разделения двух сцепленных кольцевых молекул ДНК, катализируемой ДНК-топоизомеразой типа II. Действие этих ферментов (в отличие от реакций, катализируемых ДНК-топоизомеразами типа I) сопряжено с гидролизом АТР и некоторые из них способны сообщать спирали ДНК дополнительное напряжение. ДНК-топоизомеразы типа II обнаруживаются и у прокариот, и у эукариот, по всей вероятности, они участвуют во многих реакциях, имеющих отношение к ДНК.
Топоизомераза типа II ковалентно связывается с обеими цепями двойной спирали ДНК и вносит в нее на время двухцепочечный разрыв. Ферменты этого типа активируются под действием тех участков на хромосомах, где перекрестились спирали. Присоединившись к такому перекресту, топоизомераза: 1) обратимо разрывает одну из двух двойных спиралей, создавая тем самым для другой своего рода «ворота», 2) вынуждает вторую двойную спираль пройти через этот разрыв и 3) сшивает обе разорванные цепи, а затем отделяется от ДНК. Действуя подобным образом, топоизомеразы типа II очень быстро разделяют две сцепленные кольцевые молекулы ДНК (рис. 5-53). Точно так же предотвращают они и спутывание молекул ДНК, которое в противном случае неизбежно создавало бы при репликации серьезную проблему. Известны температурочувствительные мутанты дрожжей, вырабатывающие топоизомеразу II, которая при 37°С инактивируется. Если нагреть эти дрожжевые клетки до такой температуры, то их хромосомы в процессе митоза остаются спутанными и не могут разойтись. Насколько необходим для распутывания хромосом такой «инструмент», как топоизомераза II, поймет каждый, кто хоть раз пытался распутать безнадежно запутавшуюся леску, не имея под рукой ножниц.
5-28
5.3.11. Репликация ДНК у эукариот и прокариот в основных чертах сходна [24]
Почти все, что мы знаем о репликации ДНК, удалось выяснить в опытах с очищенными мультиферментными системами бактерий и бактериофагов, способными осуществлять репликацию ДНК in vitro. Получение таких систем в 1970-х годах заметно облегчилось после того, как удалось выделить мутанты по целому ряду различных генов, ответственных за репликацию, которые можно было использовать для идентификации и очистки соответствующих белков (рис. 5-54).
У эукариот энзимология репликации ДНК пока еще детально не изучена, главным образом потому, что получать здесь необходимые мутантные формы гораздо труднее. Однако схема репликации у прокариот и эукариот в основных чертах, включая геометрию репликационной вилки и потребность в РНК-затравке, по-видимому, одинакова. Главное различие заключается в том, что у эукариот ДНК реплицируется не как таковая, а в виде хроматина, в котором она прочно связана с белками, принадлежащими к классу гистонов. В гл. 8 мы узнаем, что гистоны образуют комплексы в форме дисков, вокруг которых обвивается эукариотическая ДНК, в результате чего возникают регулярно повторяющиеся структуры, называемые нуклеосомами. Нуклеосомы располагаются вдоль молекулы ДНК с интервалами 200 пар оснований. Быть может, именно этим объясняется тот факт, что новые фрагменты отстающей цепи ДНК закладываются у эукариот с интервалами в 10 раз более короткими (от 100 до 200 нуклеотидов), чем у бактерий (от 1000 до 2000 нуклеотидов). Кроме того, если нуклеотиды служат барьерами, на время останавливающими продвижение ДНК-полимеразы, присутствие хроматина (а не голой ДНК) может, вероятно, объяснить и то, что репликационные вилки движутся у эукариот приблизительно в 10 раз медленнее, чем у бактерий.
Рис. 5-54. Получение у бактерий и бактериофагов мутантов с различными нарушениями репликации ДНК открыло возможности для выявления и очистки ферментов, выполняющих какую-либо еще не известную функцию, необходимую для репликации ДНК у прокариот. Использованные здесь температурочувствительные мутанты принадлежат к так называемым условным мутантам, обычно их фермент нормально функционирует при низкой температуре и не работает при высокой. У «безусловных» мутантов с нарушениями репликации синтез ДНК не идет ни при низкой, ни при высокой температуре, и потому эти мутанты обречены на гибель. В модифицированной форме такие «тесты на комплементацию in vitro» полезны также при биохимическом изучении многих других процессов.
Заключение
Самокорректирующая ДНК-полимераза катализирует полимеризацию нуклеотидов на обеих цепях спирали ДНК в направлении 5′ → 3′, копируя матрицу с высокой степенью точности. Поскольку две цепи двойной спирали ДНК аптипараллелъны, в направлении 5′ → 3′ может непрерывно синтезироваться лишь одна из двух цепей (ее называют ведущей). Другая, отстающая цепь синтезируется в виде коротких фрагментов по принципу «шитья назад иголкой». Самокорректирующая ДНК-полимераза не способна начинать синтез новой цепи. Поэтому для закладки фрагментов отстающей цепи ДНК используются короткие молекулы РНК-затравки, которые позже удаляются — их заменяет ДНК.
Процесс репликации ДНК требует совместного действия многих белков. В нем участвуют: 1) ДНК-полимераза и ДНК-праймаза, катализирующие полимеризацию нуклеозидтрифосфатов; 2) ДНК-геликазы и дестабилизирующие белки, помогающие расплести спираль ДНК, которую предстоит копировать; 3) ДНК-лигаза и фермент, разрушающий молекулы РНК-затравки; они нужны для сшивания прерывисто синтезируемых фрагментов отстающей цепи ДНК; 4) ДНК-топоизомеразы, помогающие решить проблемы кручения и спутывания спирали ДНК; 5) инициаторные белки, присоединившиеся к специфическим последовательностям ДНК в точке начала репликации и способствующие образованию здесь новой репликационной вилки. В точке начала репликации к ДНК-матрице сначала присоединяется белковый комплекс, состоящий из ДНК- геликазы и ДНК-праймазы (его называют праймосомой); затем к этому комплексу добавляются другие белки и возникает мультиферментный комплекс — «репликационная машина», которая и осуществляет синтез ДНК.
Транскрипция белка
Генетическая информация клеток эукариот закодирована в молекуле ДНК и располагается в ядре. ДНК не может пройти через ядерные поры так как слишком большая. В этом ей помогает специальная РНК, выступающая посредником между ядром и цитоплазмой. Эта РНК получила название матричной или информационной. В английском языке этот посредник называется более чем логично — messenger (посланник).
Что собой представляет транскрипция ДНК?
Транскрипция ДНК — это синтез мРНК, которая является комплементарной одной из цепей ДНК. Этот синтез катализируется при помощи фермента РНК-полимеразы.
Как считывается генетическая информация
Генетическая информация считывается в результате распускания небольшого фрагмента двойной спирали — это примерно один спиральный виток. мРНК комплементарно достраивается к участку ДНК. Поэтому на протяжении определенного периода времени можно наблюдать двойную спираль ДНК-РНК. Однако такая спираль не отличается хорошей устойчивостью как, к примеру, спираль ДНК-ДНК и быстро распадается.
Происходит высвобождение одноцепной РНК и восстановление двойной спирали ДНК. Вместо тимина молекулы мРНК содержат урацил. Это имеет смысл, так как биосинтез тимина требует больших затрат энергии, чем биосинтез урацила. Стабильность и точность для молекул РНК не так важна — они находятся и функционируют в клетке меньшее количество времени.
Информация при транскрипции считывается в таком же направлении, что и при репликации.
Синтез молекулы мРНК происходит в направлении 5r-3r. Из ДНК информация считывается в направлении 3r-5r.
ДНК-зависимая РНК-полимераза — главный фермент транскрипции. Если речь идет о прокариотах, то в процессе участвует один фермент, если об эукариотах — комплекс из трех ферментов. Этому ферменту отводится роль катализатора синтеза РНК.
Молекулы синтезированной РНК-полимеразы на матрице ДНК — это первичный транскрипт. Смесь из таких молекул получила название гетерогенной ядерной РНК (гяРНК).
Зрелая мРНК — молекула, служащая матрицей для синтеза белка.
РНК-полимераза
РНК-полимераза является сложным ферментом с четвертичной структурой. У нее есть несколько субъединиц: это две а, одна b, одна br, одна s.
Молекулярная масса фермента — 500 кДа.
Чтобы РНК-полимераза работала нормально, важно наличие нескольких элементов:
- матрицы;
- активированные предшественники;
- двухвалентные ионы.
ДНК — наиболее эффективная матрица для РНК-полимеразы, к которой фермент достраивает комплементарную копию. В качестве активированных предшественников в этом случае используются 5r-рибонуклеозидтрифосфаты.
Чтобы транскрипция случилась, нужны четыре трифосфата: АТФ, ТТФ, УТФ, ГТФ. Активность фермента PH K-полимеразы обеспечивается за счет наличия двухвалентных ионов Mg₂ и Mn₂.
Указанные выше факторы говорят о том, что РНК-полимераза и ДНК-полимераза близки по своим характеристикам. Тем не менее можно выделить и существенные различия:
- РНК-полимераза не нуждается в затравке. Синтез может начинаться с новой цепи. Если говорить о ДНК-полимеразе, то здесь важно наличие цепи со свободной OH-группой в положении С;
- при транскрипции ДНК-матрица сохраняется в полной мере. В случае репликации происходит сохранение только одной «старой» цепи. По этой причине можно говорить о полуконсервативном характере репликации. Для РНК-полимеразы не свойственна эндонуклеазная активность;
- ДНК-полимераза контролирует синтез новой цепи на правильность — ошибочные нуклеотиды вырезаются. Для РНК-полимеразы контроль правильности синтеза цепи не характерен. По этой причине при транскрипции есть вероятность ошибок: 1:150 нуклеотидов. Это в десять тысяч раз больше, чем в случае репликации.
Этапы процесса транскрипции
Процесс транскрипции отличается сложностью. Выделяют несколько его этапов:
- Инициация. На этом этапе РНК-полимераза присоединяется к началу участка ДНК (с нее считывается информация).
- Элонгация. Этап характеризуется построением цепи мРНК.
- Терминация. Синтез заканчивается, первичный транскрипт высвобождается.
- Процессинг. На этом этапе происходят изменения новосинтезированной молекулы мРНК или первичным транскриптом. Эти изменения происходят до ее участия в биосинтезе белка.
Все этапы транскрипции связаны с соответствующими участками ДНК. Участвующая в транскрипции часть ДНК включает отдельные обязательные фрагменты.
Участок ДНК, распознаваемы РНК-полимеразой — промотор.
Он находится прямо перед участком, с которого начинается транскрипция. Размер этого участка — примерно 40 пар оснований. Промоторные участки в разных организмах имеют одинаковое строение.
В ходе исследований, проводимых на промоторах E coli, было установлено, что полимераза способна распознавать на молекуле ДНК две нуклеотидные последовательности из 6 нуклеотидов. Они разделены примерно 25 нуклеотидами.
Фрагмент ДНК, из которого начинается синтез, мРНК называется лидером.
Согласно исследованиям, синтез мРНК берет начало с аденозитрифосфата или гианозитрифосфата, более известных как АТФ и ГТФ.
В результате новообразованная мРНК имеет получает трифосфатную группу на 5r-конце и группу OH на 3r-конце.
Весь участок ДНК при помощи которого происходит синтез мРНК, называется цистроном или единицей транскрипции.
Особенность транскрипции заключается в следующем: процесс считывания генетической информации с молекулы ДНК осуществляется только с одной, при этом любой, цепи двойной спирали.
Антипараллельный участок цепи не содержит генетической информации. Основная его функция — поддерживать целостность участка ДНК.
Участок ДНК с расположенным на нем сигналом, указывающим на окончание процесса транскрипции — это терминатор или трейлер.
В состав терминатора входят гуанин-цитозин и аденин-тимин обогащенные участки. Участки вдоль одной цепи являются зеркально симметричными, и могут быть запертыми на самих себя — таким образом они создают «шпильку».
Эта шпилька препятствует движению фермента и останавливает процесс транскрипции. Процессинг или созревание — это превращение первичного транскрипта в зрелую мРНК.
У прокариот и эукариот процессинг существенно различается. Это связано со спецификой строения генома. У прокариотических клеток первичный транскрипт может содержать информацию не только об одной полипептидной цепи, но и о нескольких соседних, а также о РНК.
В этих клетках процессинг включает этап, связанный с разрезанием первичного транскрипта на несколько составных частей — если такие имеются.
Если транскрипт включает информацию только об одной полипептидной цепи, то процесс биосинтеза иногда начинается до того, как транскрипция закончится.
Экзоны или информационные участки в геноме эукариот характеризуются чередованием с неинформационными участками (интронами). По этой причине информация об отдельном полипептиде характеризуется большей физической длиной. В первичном транскрипте содержатся экзоны и интроны, относящиеся к одной полипептидной цепи.