Механизмы исправления ошибок во время репликации ДНК и ее репарация вследствие повреждений на протяжении всего жизненного цикла клетки.
Основные моменты:
-
Клетки имеют различные механизмы предотвращения возникновения мутаций – необратимых изменений в ДНК
-
В процессе синтеза ДНК, большинство ДНК-полимераз «проверяют свою работу» и проводят замену бо́льшей части ошибочно вставленных нуклеотидов. Этот процесс можно назвать исправлением ошибок.
-
Сразу после синтеза ДНК любые оставшиеся ошибочные нуклеотиды обнаруживаются и заменяются в так называемом процессе репарации ошибочно спаренных нуклеотидов.
-
Если ДНК повреждена, она может быть восстановлена с помощью различных механизмов, например, путём прямой репарации, эксцизионной репарации или путём восстановления двухцепочечных разрывов
- пострепликативной репарации.
Введение
Как ДНК связана с раком? Рак возникает при неконтролируемом делении клеток, когда игнорируются клеточные «стоп»-сигналы, что приводит к образованию опухоли. Это неправильное поведение клеток вызвано накопившимися мутациями — необратимыми изменениями последовательности ДНК клетки.
На самом деле, ошибки в процессе репликации и повреждения ДНК возникают в клетках нашего тела постоянно. Однако в большинстве случаев они не приводят к раку и даже не вызывают мутаций, такие ошибки обычно обнаруживаются и исправляются в процессе репарации ДНК. Если же повреждение исправить не удаётся, то в клетке включается механизм самоуничтожения — (апоптоз), который предотвращает передачу поврежденной ДНК дочерним клеткам.
Мутации возникают и передаются дочерним клеткам только тогда, когда эти механизмы не справляются. В частности, рак возникает в случае накопившихся в одной клетке мутаций генов, связанных с делением.
В этой статье мы подробно рассмотрим механизмы, используемые клетками для исправления ошибок, которые возникают в процессе репликации. К ним относятся:
-
Исправление ошибок – процесс, который возникает во время репликации ДНК.
-
Репарация ошибочно спаренных нуклеотидов, которая происходит сразу же после репликации ДНК.
-
Механизмы репарации, которые выявляют и исправляют повреждения ДНК на протяжении всего клеточного цикла
Исправление ошибок
ДНК-полимеразы — это ферменты, участвующие в репликации ДНК. Во время копирования ДНК большинство ДНК-полимераз «проверяют», корректный ли нуклеотид они добавляют. Этот процесс называется исправлением ошибок. Если полимераза обнаружит, что был добавлен неправильный нуклеотид, она сразу же удалит и заменит его и только после этого продолжит синтез ДНКstart superscript, 1, end superscript.
Репарация ошибочно спаренных нуклеотидов
Процесс исправления избавляет от основной массы ошибок, но не от всех. После создания новой ДНК запускается механизм репарации ошибочно спаренных нуклеотидов — удаления и замены ошибочно спаренных нуклеотидов, оставшихся в результате репликации. Исправление несоответствий между парами оснований также может включать в себя исправление небольших вставок и делеций, возникающих вследствие «соскальзывания» полимеразы с исходной цепи squared.
Как происходит восстановление неправильно спаренных нуклеотидов? Во-первых, белковый комплекс распознаёт неправильно спаренный нуклеотид и связывается с ним. Другой комплекс разрезает ДНК в области несовпадения, а ещё одна группа ферментов отщепляет некорректный нуклеотид вместе с небольшим участком вокруг него. Затем ДНК-полимераза заполняет этот пробел правильными нуклеотидами, а фермент ДНК-лигаза сшивает разрывы в цепиsquared.
Удивительно: как белки, участвующие в восстановлении ДНК, определяют, «кто прав» во время репарации ошибочно спаренных нуклеотидов? То есть, когда два основания неправильно соединены (как G (гуанин) и T (тимин) на рисунке выше), какое из этих двух оснований должно быть удалено и заменено?
У бактерий можно отличить исходную и дочернюю цепи ДНК по метилированным основаниям. На исходной цепи ДНК есть метильные (minus, start text, C, H, end text, start subscript, 3, end subscript) группы, присоединенные к некоторым из ее оснований, а у дочерней цепи таких групп еще нетcubed.
У эукариот процессы, позволяющие идентифицировать исходную цепь при устранении несоответствий, включают распознавание одноцепочечных разрывов, которые обнаруживаются только у дочерней цепи cubed.
Механизмы репарации ДНК
С ДНК может что-нибудь случиться практически в любой момент жизни клетки, а не только во время репликации. Фактически, ДНК постоянно повреждается из-за воздействия внешних факторов: ультрафиолетового излучения и радиации, химических веществ, не говоря уже о спонтанных процессах, которые протекают даже без вмешательства окружающей среды!start superscript, 4, end superscript
К счастью, наши клетки имеют механизмы восстановления, с помощью которых они находят и исправляют большинство повреждений ДНК. Можно выделить несколько типов репарации:
-
Прямая репарация. Некоторые повреждения ДНК, вызванные химическими реакциями, могут быть «исправлены» находящимися в клетке ферментами.
-
Эксцизионная репарация. Повреждение одного или нескольких нуклеотидов ДНК часто исправляется удалением и заменой поврежденного участка. При эксцизионной репарации оснований удаляется только поврежденное основание. В случае эксцизионной репарации нуклеотидов, как и в случае репарации ошибочно спаренных нуклеотидов, которое мы рассмотрели выше, удаляются целиком нуклеотиды.
-
Репарация двухцепочечных разрывов: Существуют два основных способа: негомологичное соединение концов и гомологичная рекомбинация. Они используются для восстановления двухцепочечных разрывов ДНК (когда вся хромосома разделяется на две части).
Прямая репарация
В некоторых случаях клетка может исправить повреждение ДНК, обратив вызвавшую его реакцию. Дело в том, что «повреждение ДНК» — это, как правило, присоединение к ней лишней группы в результате химической реакции.
Например, гуанин (G) может подвергаться реакции с присоединением метильной (minus, start text, C, H, end text, start subscript, 3, end subscript) группы к атому кислорода в азотистом основании. Если это не исправить, метил-содержащий гуанин будет связываться с тимином (Т), а не с цитозином (С) во время репликации ДНК. К счастью, у людей и многих других организмов есть фермент, который может удалить метильную группу, обратив реакцию, и тем самым вернуть азотистое основание в нормальное состояниеstart superscript, 5, end superscript.
Эксцизионная репарация оснований
Эксцизионная репарация оснований — это механизм, используемый для обнаружения и удаления определенных типов поврежденных азотистых оснований. Ключевую роль в нем играет группа ферментов, называемых гликозилазами. Каждая гликозилаза обнаруживает и удаляет определенный вид поврежденных оснований.
Например, в процессе реакции дезаминирования цитозин может превратиться в урацил — основание, обычно встречающееся только в РНК. Во время репликации ДНК урацил будет соединяться с аденином, а не с гуанином (в отличие от цитозина), поэтому такое превращение может привести к возникновению мутацииstart superscript, 5, end superscript.
Для предотвращения подобных изменений гликозилаза, являющаяся частью сигнального пути эксцизионной репарации, обнаруживает и удаляет дезаминированные цитозины. После того, как основание было удалено, удаляется и оставшаяся часть нуклеотида, а другие ферменты заполняют пробелstart superscript, 6, end superscript.
Эксцизионная репарация нуклеотидов
Эксцизионная репарация нуклеотидов — это еще один способ удаления и замены поврежденных оснований. В результате нее обнаруживаются и корректируются повреждения, которые искажают форму двойной спирали ДНК. Например, азотистые основания могут измениться, присоединив к себе громоздкие группы атомов, в частности, в результате воздействия химических веществ, содержащихся в сигаретном дымеstart superscript, 7, end superscript.
Эксцизионная репарация нуклеотидов также используется для устранения повреждений, вызванных ультрафиолетовым излучением, например, при получении солнечного ожога. Под воздействием УФ-излучения цитозин и тимин могут вступать в реакцию с соседними основаниями, которые также являются цитозином или тимином, образуя при этом связи, изменяющие форму двойной спирали и вызывающие ошибки в процессе репликации ДНК. Наиболее распространенный тип таких связей — тиминовый димер — он состоит из двух тиминовых оснований, вступающих в реакцию друг с другом и образующих химическую связьstart superscript, 8, end superscript.
При эксцизионной репарации нуклеотидов поврежденные нуклеотиды удаляются вместе с соседними нуклеотидами. В этом процессе хеликаза (фермент, раскручивающий ДНК) раскрывает ДНК, образуя пузырь, а ферменты, разрезающие ДНК, отсекают поврежденную часть пузыря. Полимераза заполняет пробел, а лигаза сшивает разрыв в цепиstart superscript, 9, end superscript.
Репарация двухцепочечных разрывов
Некоторые факторы окружающей среды, например, радиация, могут вызывать разрывы обеих цепочек ДНК (разделение хромосомы на две части). Такие повреждения ДНК, если верить комиксам, ведут к появлению супергероев, но могут встречаться и после реальных катастроф, например, Чернобыльской.
Двухцепочечные разрывы опасны, потому что большие сегменты хромосом и сотни содержащихся в них генов могут быть потеряны, если разрыв не будет восстановлен. Существует два способа восстановления двухцепочечных разрывов ДНК: негомологичное соединение концов и гомологичная рекомбинация.
При негомологичном соединении концов два разорванных конца хромосомы просто склеиваются обратно. Этот механизм восстановления является «грубым» и неточным, в результате в месте разрыва, как правило, либо теряются нуклеотиды, либо добавляются лишние, что может привести к мутациям. Но это в любом случае лучше потери целого фрагмента хромосомыstart superscript, 10, end superscript.
При гомологичной рекомбинации для восстановления разрыва используется фрагмент из гомологичной хромосомы, который соответствует поврежденной хромосоме (или из сестринской хроматиды, если ДНК была реплицирована). В этом процессе две хромосомы объединяются, и неповрежденная область гомологичной хромосомы или хроматиды используется в качестве матрицы для замены поврежденной области. Гомологичная рекомбинация работает «чище», точнее, чем негомологичное соединение концов, и обычно не приводит к образованию мутацийstart superscript, 11, end superscript.
Репарация ДНК и заболевания человека
Доказательства важности механизмов репарации получены на основе генетических заболеваний человека. Во многих случаях мутации в генах, которые кодируют белки, участвующие в репарации, связаны с наследственным раком. Например:
-
Наследственный неполипозный колоректальный рак (также называемый синдромом Линча) вызван мутациями в генах, кодирующих белки, которые участвуют в репарации ошибочно спаренных нуклеотидовstart superscript, 12, comma, 13, end superscript. Поскольку такие нуклеотиды не восстанавливаются, у людей, страдающих этим синдромом, мутации накапливаются гораздо быстрее, чем у здоровых. Это может привести к развитию опухолей толстой кишки.
-
Люди с пигментной ксеродермой очень чувствительны к ультрафиолетовому излучению. Это вызвано мутациями в белках, участвующих в эксцизионной репарации нуклеотидов. Когда они не функционируют, димеры тимина и другие виды повреждений, вызванные ультрафиолетовым излучением, перестают восстанавливаться. У людей с пигментной ксеродермой после нескольких минут пребывания на солнце могут возникнуть сильные солнечные ожоги, и около половины из них заболевают раком кожи в возрасте до 10 лет, если только они не избегают солнечных лучейstart superscript, 14, end superscript.
ДНК-полимераза — фермент,
участвующий в репликации
ДНК.
Ферменты этого класса катализируют
полимеризацию дезоксирибонуклеотидов вдоль
цепочки нуклеотидов ДНК,
которую фермент «читает» и использует
в качестве шаблона. Тип нового нуклеотида
определяется по принципу
комплементарности с
шаблоном, с которого ведётся считывание.
Собираемая молекула комплементарна
шаблонной моноспирали и идентична
второму компоненту двойной спирали.
Выделяют ДНК-зависимую
ДНК-полимеразу (КФ 2.7.7.7),
использующую в качестве матрицы одну
из цепей ДНК, и РНК-зависимую
ДНК-полимеразу (другое
название обратная
транскриптаза, КФ 2.7.7.49),
способную также к считыванию информации
с РНК (обратная
транскрипция).
ДНК-полимеразу
считают холоферментом,
поскольку для нормального функционирования
она требует присутствия ионов магнияв
качестве кофактора. В отсутствие ионов
магния о ней можно говорить как
об апоферментe.
ДНК-полимераза
начинает репликацию ДНК, связываясь с
отрезком цепи нуклеотидов. Среднее
количество нуклеотидов, присоединяемое
ферментов ДНК-полимеразой за один акт
связывания/диссоциации с матрицей,
называют процессивностью.
Действие
ДНК-полимеразы
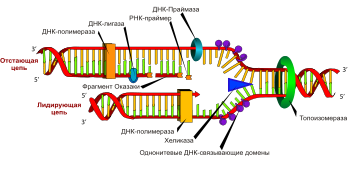
Репликация
ДНК
Как
известно, две цепи молекулы ДНК
антипараллельны. Разные концы одной
цепи называются 3’-конец и 5’-конец.
Репликация происходит путем непрерывного
роста нуклеотида за нуклеотидом обеих
новых цепей одновременно. Матрица
считывается ДНК-полимеразой только в
направлении 3’-5’, добавляя свободные
нуклеотиды к 3’-концу собираемой цепочки.
Поэтому синтез ДНК происходит непрерывно
только на одной из матричных цепей,
называемой «лидирующей».
Во второй цепи («отстающей»)
синтез происходит короткимифрагментами.
Ни
одна из известных ДНК-полимераз не может
создать цепочку «с нуля»: они в состоянии
лишь добавлять нуклеотиды к уже
существующей 3’-гидроксильной группе.
По этой причине ДНК-полимераза нуждается
впраймере,
к которому она могла бы добавить первый
нуклеотид. Праймеры состоят из
оснований РНК и
ДНК, при этом первые два основания всегда
РНК-основания. Праймеры синтезируются
другим ферментом — праймазой.
Ещё один фермент — геликаза —
необходим для раскручивания двойной
спирали ДНК с формированием одноцепочечной
структуры, которая обеспечивает
репликацию обеих цепочек в соответствии
с полуконсервативной моделью репликации
ДНК.
Некоторые
ДНК-полимеразы обладают также способностью
исправлять ошибки во вновь собираемой
цепочке ДНК. Если происходит обнаружение
неправильной пары нуклеотидов,
ДНК-полимераза откатывается на один
шаг назад. Благодаря своей
3′-5′ экзонуклеазной гидролитической
активности ДНК-полимераза может исключить
неправильный нуклеотид из цепочки и
затем вставить на его место правильный,
после чего репликация продолжается в
нормальном режиме.
Многообразие
ДНК-полимераз
Структура
ДНК-полимераз достаточно жёстко
фиксирована. Их каталитические субъединицы
очень мало различаются в различных
видах живых клеток. Такая фиксация
структуры обычно появляется там, где
отсутствие разнообразия обусловлено
огромной важностью или даже незаменимостью
для функционирования клетки.
Генами
некоторых вирусов тоже
кодируются особые ДНК-полимеразы,
которые могут избирательно реплицировать
вирусные ДНК. Ретровирусы обладают
геном необычной ДНК-полимеразы, называемой
ещё обратной
транскриптазой,
являющейся РНК-зависимой ДНК-полимеразой
и осуществляющей сборку ДНК на основе
шаблонной РНК.
Семейства
ДНК-полимераз
На
основании своей структуры ДНК-полимеразы
могут быть разбиты на семь различных
семейств: A, B, C, D, X, Y, и RT.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
рис.1 Модифицированные азотистые основания ДНК, удаляемые ДНК–гликозилазами: а – урацил; б – гипоксантин; в – 5–гидроксицитозин; г – 2,5-диамино-4-формамидопиримидин; д – 7,8-дигидро-8-оксогуанин; е – мочевина; ж – тимингликоль; з – 5-формилурацил; и – 5-гидроксиметилурацил; к – 3-метиладенин; л – 7-метилгуанин; м – 2-метилцитозин [Партушев, 2000].
АP-эндонуклеаза создает ник (одноцепочечный разрав) с 3’-ОН и 5’-dRP концами. Для большинства млекопитающих характерен
тип BER репарации с включением одного нуклеотида. В этом случае ДНК-полимераза β вставляет 1 нуклеотид в 3’-конец праймера и затем удаляет 5’-dRP фрагмент с помощью своей dRP-лиазной активности. Получившийся ник сшивается ДНК-лигазой. Альтернативным является путь так называемой длинно-заплаточной BER репарации. Он реализуется в тех случаях, когда 5’-dRP фрагмент модифицирован и не может быть удален с помощью ДНК–полимеразы β. В этом случае Pol β проводит синтез ДНК с вытеснением запирающей цепи, 2 — 13 нуклеотидов. Образующийся в результате свисающий 5’-конец ДНК выщепляется флэпэндонуклеазой FEN1. Pol бета, Pol дельтаи Pol епсилон могут вести ситнез ДНК во время длинно-заплаточной репарации. Идентичность полимераз, вовлеченных в этот процесс in vitro, еще не ясна, но показано, что Pol β всегда инициирует синтез ДНК.
Зависимость того, какой из путей BER реализуется, определяется бифункциональными ДНК-гликозилазами, которые имеют дополнительную
АP-лиазную активность. При комбинации ДНК-гликозилазной, АP-лиазной и АP-эндонуклеазной активностей внутри одного фермента в поврежденной цепи ДНК образуется однонуклеотидная брешь с 3’-ОН и 5’-P концами, которую может застроить Pol β.
Дополнительная сложность в понимании механизмов переключения внутри BER состоит в том, что две новые полимеразы Pol i и Pol лямбда также имеют dRP-лиазнуюактивность. Таким образом, можно предположить, что они, как и Pol бета, являются участниками тех процессов репарации, где необходимо выщепление dRP фрагмента. Pol лямбда является близким гомологом Pol бета со сходными ферментативными свойствами. Также как и Pol бета, она лишена 3’—>5’экзонуклеазной активности и имеет низкую процессивность синтеза на частичном ДНК-дуплексе, содержащем свисающий 5’-участок матрицы, процессивный синтез возможен в брешах с 5’-P концами. Таким образом, Pol лямбда является подходящим кандидатом для BER синтеза. Более того,
Pol лямбда заменяет Pol бета в реконструированных BER системах, репарирующих урацил-содержащие ДНК, iv vitro. Однако, клетки мышей Pol лямбда -/- не чувствительны к обработке перекисью или метилметансульфонатом, агентам, которые, помимо других типов повреждений, продуцируют АР-сайты и окисленные формы оснований. Отсюда можно заключить, что Pol лямбда не является необходимой для BER в клетках, где есть, Pol дельта и Pol епсилон. Интересно, что Pol лямбда может эффективно процессировать ДНК при очень низкой концентрации dNTP (около 1 мкМ), что может означать ее участие в любых клеточных процессах в фазе G0 при низкой концентрации dNTP. В поддержку этой гипотезы выступает тот факт, что экспрессия Pol лямбда зависит от клеточного цикла, и наибольшее количества белка экспрессируется при переходе из S- в М-фазу и в спокойных клетках.
Pol i принадлежит к Y семейству полимераз. Основная функция полимераз этого семейства – утилизация ДНК-повреждений, блокирующих работу репликативных ДНК-полимераз. Однако,
исходя из некоторых свойств Pol i, можно предположить, что этот фермент является участником альтернативного процесса BER репарации. Pol i обладает низкой поцессивностью, лишена 3’>5’экзонуклеазной активности и способна застраивать брешь в 1 — 5 нуклеотидов iv vitro. Pol i может замещать Pol бета в реконструированных системах BER, репарирующих урацил-содержащие ДНК, iv vitro, благодаря наличию dRP-лиазной и ДНК-полимеразной активностей. Кинетические исследования реакции нуклеотидного встраивания овыявили дополнительные данные, свидетельствующие в пользу того, что Pol i может принимать участие в репарации. Pol i вставляет dTMP напротив А в ДНК-матрицу с большей эффективностью, чем любой другой дезоксинуклеозидмонофосфат, при этом точность ДНК-синтеза сопоставима с параметрами для Pol бета. Основываясь на этих данных, можно предположить, что Pol i – участник BER репарации оснований уридина, который образуется после встраивания dUMP напротив А во время ошибочной репликации. Pol i также
может вставлять dGМP напротив Т со скоростями, близкими к скорости включения корректного нуклеотида. Более того, на матрице, содержащей 2 или более последовательных Т, вторым встраиваемым нуклеотидом оказался dGMP. Подобные результаты служат основой для гипотезы, что Pol i может быть участником альтернативной BER репарации в тех слсучаях, когда dG был ошибочно удален гликозилазой из G-T или G-U некомплементарной пары, образовавшейся в результате дезаминации 5-метилцитозина или цитозина. Возможная роль Pol i в трансляции синтеза ДНК и процессе соматических гипермутаций рассмотрена ниже.
Возможность участия пяти ядерных полимераз в BER, три из которых обладают dRP-лиазной активностью, была мало изучена в клетках и на модельных животных, лишенных более чем одной полимеразы. Поэтому данные о субстратной специфичности и взаимодействии полимеразо-акцепторных белков, необходимых в BER, недостаточны. Помимо BER репарации, Pol бетта принимает участие в репарации одноцепочечных разрывов; функционирование
этого процесса нарушено у пациентов с наследственной спинномозговой атаксией. Одноцепочечные разрывы генерируются эндогенными или экзогенными агентами. Такие разрывы часто содержат однонуклеотидные бреши с 3’ и/или 5’ модифицированными концами, то есть очень похожи на субстраты BER. Так вот, интересно посмотреть, способны ли другие полимеразы с подобным набором исходных данных (Pol i и Pol лямбда) принимать участие в репарации одноцепочечных разрывов.
Четвертой ДНК-полимеразой в клетках человека, обладающей dRP-лиазной активностью, является митохондриальная ДНК-полимераза гамма. Pol гамма – единственная ДНК-полимераза, обнаруженная в митохондриях, следовательно, она ответственна за все преобразованиях ДНК, происходящие в этой органелле. Митохондрия является объектом интенсивного повреждения ДНК активными формами кислорода, генерируемыми во время окислительного фосфорилирования. Эти повреждения эффективно репарируются набором митохондриальных белков, которые включают в себя АP-эндонуклеазу, Pol гамма,
мтДНК-лигазу. Сам процесс репарации сходен с однонуклеотидным процессом BER в ядерной ДНК.
Таблица. ДНК-гликозилазы и эндонуклеазы клеток микроорганизмов и человека, участвующие в BER.
В универсальном механизме эксцизионной репарации как прокариоты, так и эукариоты гидролизуют 3–5-ю фосфодиэфирную связь с 3′-конца отповреждения. При этом прокариоты гидролизуют также 8-ю связьот 5’-конца измененного нуклеотида, тогда как у эукариотических организмовпроисходит одноцепочечный разрыв на расстоянии 21–25 нуклеотидов отповреждения со стороны его 5’-конца. Таким образом, прокариоты удаляютизмененный нуклеотид в составе 12–13-членных олигомеров, тогда как
эукариоты – в составе одноцепочечных фрагментов ДНК длиной в 27–29 нуклеотидов. Ферментная система, вносящая такие двойные одноцепочечные
разрывы, получила название эксцизионной нуклеазы (эксцинуклеазы). Образующаяся в молекуле репарируемой ДНК одноцепочечная брешь далее заполняется с помощью ДНК-полимеразы, а фосфодиэфирная связь в остающемся одноцепочечном разрыве восстанавливается ДНК-лигазой.
NER устраняет ДНК-повреждения посредством вырезания олигонуклеотида, содержащего это повреждение, с образованием бреши в ДНК размером ~ 30 нуклеотидов. Эта брешь застраивается одной или двумя полимеразами В семейства Pol дельта или Pol епсилон. Любая из этих ДНК-полимераз способна застраивать подобную брешь в реконструированной системе, содержащей очищенные белки млекопитающих, in vitro. При изучении NER в системе фибробластов человека с нарушенной проницаемостью и в ядерном экстракте HeLa клеток обнаружили, что обе полимеразы необходимы для восстановления ДНК после УФ-облучения. Исследования
в дрожжевых системах показали, что та или другая ДНК-полимераза существенно дополняют репарацию УФ-поврежденной ДНК. Более поздние исследования показали, что оба фермента необходимы для эффективной NER репарации в дрожжевых экстрактах. Pol дельта и Pol епсилон способны вести процессивный синтез ДНК при наличии дополнительного фактора PCNA, который «надевается» на ДНК с помощью пятисубъединичного комплекса RFC. Репарационный синтез как Pol дельта, так и Pol епсилон также требует присутствия этих факторов процессивности.
Эксцизионная репарация нуклеотидов: введение
ЭР — эксцизионная репарация ДНК; ЭР-комплекс включает ПАРП , XRCC1 , ДНК-лигазу III и ДНК-полимеразу бета .
Два пути осуществления NER:
1. Гидролиз фосфодиэфирной связи по 3′- или 5′- концу на некотором расстоянии от ошибочно спаренного (поврежденного) нуклеотида, который далее целиком удаляется под действием 5′->3′- (или 3′->5′-) экзонуклеазы, гидролизующей
цепь ДНК нуклеотид за нуклеотидом в соответствующем направлении от первоначального одноцепочечного разрыва в репарируемой ДНК. Образующаяся брешь далее заполняется ДНК-полимеразой. Такой механизм репарации реализуется у E. coli и человека для вырезания неповрежденных (немодифицированных) ошибочно спаренных нуклеотидов. Механизм последовательного эндо- и экзонуклеазного расщепления ДНК не используется для удаления поврежденных (измененных) нуклеотидов. Это связано с тем, что такие нуклеотиды часто являются ингибиторами экзонуклеаз.
2. Функционирует у всех исследованных видов организмов и заключается в использовании ферментной системы, которая вносит одноцепочечные разрывы по обе стороны от поврежденного нуклеотида на некотором расстоянии от него с последующим удалением одноцепочечного фрагмента ДНК, содержащего измененный нуклеотид.
Гидролизуется 3-5-фосфодиэфирную связь с 3′-конца от повреждения. При этом прокариоты гидролизуют также 8-связь от 5′-конца
измененного нуклеотида, тогда как у эукариотических организмов происходит одноцепочечный разрыв на расстоянии 21-25 нуклеотидов от повреждения со стороны его 5′-конца. Таким образом, прокариоты удаляют измененный нуклеотид в составе 12-13-членных олигомеров, тогда как эукариоты — в составе одноцепочечных фрагментов ДНК длиной в 27-29 нуклеотидов. Ферментная система, вносящая такие двойные одноцепочечные разрывы, получила название эксцизионной нуклеазы (эксцинуклеазы) . Образующаяся в молекуле репарируемой ДНК одноцепочечная брешь далее заполняется с помощью ДНК-полимеразы, а фосфодиэфирная связь в остающемся одноцепочечном разрыве восстанавливается ДНК-лигазой.
В отличие от ЭРО и прямых реверсий, которые специфичны к достаточно узкому кругу повреждений ДНК, система ЭРН, хотя и с разной эффективностью, удаляет все возможные повреждения, и потому роль ее в поддержании стабильности генома велика. ЭРН детально изучена у E. coli и активно изучается в клетках
дрожжей и человека, причем в последних благодаря раскрытию генетической природы таких заболеваний, как пигментная ксеродерма ( ХР ), синдром Кокейна ( СS ) и заболевания трихотиодистрофия ( ТТD)
В ЭРН задействовано 6-8 генов у E. coli и до 30 у человека.
Если повреждение в транскрибируемой (матричной) нити ДНК задерживает продвижение РНК-полимеразы, то специальный белковый фактор TRCF ( transcription-repair coupling factor ) сталкивает РНК-полимеразу и связывает с этим местом комплекс ферментов репарации. См. Сопряжение транскрипции и репарации ДНК у E/coli.
Ферментный ансамбль осуществляющий первые 3 стадии процесса NER называется эксцинуклеазой.
В системе эксцизионной репарации ДНК путем удаления нуклеотидов поврежденные азотистые основания вырезаются в составе олигонуклеотидов.
NER: регуляция
Для клеток животных не характерен SOS-ответ, свойственный клеткам E. coli и представляющий собой суммарную реакцию бактериальной клетки на повреждение ДНК различными агентами,
проявляющийся в усилении транскрипции генов NER. Посттрансляционные модификации белков репарации, происходящие в ответ на повреждение ДНК, не влияют на активность эксцинуклеазы человека.
Обнаружено, что повреждения ДНК стабилизирует белок р53- белок-супрессор опухолевого роста, являющийся регулятором транскрипции. Имеются данные о том, что белок р53 может взаимодействовать с белками XPB и RPA, необходимыми для NER. Однако клетки с инактивированными генами р53 (p53(-/-)), как и клетки дикого типа, эффективно удаляют из поврежденной ДНК два основных фотопродукта, возникающих под действием УФ-света, и обладают такой же устойчивостью к УФ. Поэтому считается, что белок р53 не оказывает прямого влияния на NER. Белки Cdk7 и циклин H, которые образуют Cdk-активирующую киназу, входят в состав комплекса TFIIH, что позволяет предполагать наличие связи репарации ДНК с фазами клеточного цикла.
ЭРН (NER): сопряжение с транскрипцией (ТЭРН)
Транскрибируемые последовательности нуклеотидов ДНК, особенно
в матричной цепи, репарируются с большей скоростью, чем нетранскрибируемые последовательности. В клетках больных с синдромом Кокайна не наблюдается такой асимметрии в репарации.
В клетках E. coli белковый фактор, кодируемый геном mfd и сопрягающий транскрипцию и репарацию, замещает остановившиеся перед повреждением молекулы РНК-полимеразы, что приводит к диссоциации транскрипционного комплекса. При этом он привлекает экзонуклеазный репаративный комплекс к поврежденному участку ДНК. См. Сопряжение транскрипции и репарации ДНК (ТЭРН) у E/coli
В клетках животных ген CSB кодирует белок с молекулярной массой 160 кДа, который содержит хеликазный домен и, возможно, выполняет те же функции, что и белок Mfd у E. coli . На основе поведения клеток с мутантными генами белков CSA и CSB разработана модель механизма, с помощью которого обеспечивается асимметричная репарация цепей ДНК. В соответствии с этой моделью РНК-полимераза II , остановившаяся в процессе транскрипции
перед поврежденным участком ДНК, распознается комплексом CSA-CSB и перемещается в сторону от повреждения без разрушения четвертичной структуры транскрипционного комплекса. Одновременно комплекс CSA-CSB привлекает компоненты репаративной системы XPA и TFIIH к месту повреждения ДНК и помогает сборке эксцинуклеазного комплекса. Нуклеотиды поврежденной цепи вырезаются, и брешь репарируется. После этого РНК-полимераза в составе транскрипционного комплекса продолжает транскрипцию.
NER: механизм
Процесс NER можно разделить на четыре этапа: а) распознавание поврежденного участка ДНК; б) двойное надрезание (инцизия) цепи ДНК по обеим сторонам поврежденного участка и его удаление (эксцизия); в) заполнение бреши в процессе репаративного синтеза; г) лигирование оставшегося одноцепочечного разрыва ДНК.
NER человека распознает и удаляет одиночные ошибочно спаренные нуклеотиды, а также петли длиной в 1-3 нуклеотида. NER человека способна различать цепи ДНК в случае распознавания поврежденных
нуклеотидов. Показано, что при наличии в ДНК димеров тимина циклобутанового типа вырезание нуклеотидов происходит исключительно из поврежденной цепи. Механизм такого распознавания в настоящее время неизвестен, как и молекулярный механизм узнавания самих поврежденных оснований. Система способна распознавать повреждения как сильно, так и слабо деформирующие вторичную структуру ДНК. При этом не обнаружена линейная зависимость между коэффициентом специфичности нуклеазы (kcat/km) и уровнем деформации двойной спирали ДНК. Показано, что в процессе распознавания участвуют белковые комплексы XPA/RPA, которые преимущественно связываются с поврежденной ДНК, и TFIIH, обладающий АTP-зависимой ДНК-расплетающей активностью. Последний взаимодействует с поврежденным участком ДНК и по аналогии с соответствующим механизмом у E. coli локально раскручивает ДНК, создавая основной преинцизионный комплекс с поврежденной ДНК.
Установлено, что три фермента репарации, обладающие узкой
субстратной специфичностью: ДНК-фотолиаза (удаление пиримидиновых димеров), урацилгликозилаза (удаление урацила из ДНК) и экзонуклеаза III (гидролиз ДНК в AP-сайтах), втягивают поврежденный участок из двойной спирали в полость фермента, что приводит кофактор или аминокислотные остатки активного центра этих ферментов в непосредственный контакт с расщепляемыми связями ДНК. Не исключено, что система эксцинуклеазы действует таким же образом.
Основные этапы функционирования NER, следующие за распознаванием поврежденного участка ДНК, представлены на рис. I.59. После связывания комплекса XPA-RPA с измененным участком ДНК, XPA взаимодействует с комплексом TFIIH, который создает преинцизионный комплекс, что сопряжено с гидролизом ATP. ATP-зависимое расплетание ДНК комплексом TFIIH подготавливает ее к взаимодействию с двумя XP-белками, обладающими нуклеазной активностью. XPG связывается с TFIIH и вносит одноцепочечный разрыв с 3′-конца повреждения. Аналогично комплекс ERCC1-XPF
взаимодействует с XPA в составе преинцизионного комплекса и способствует одноцепочечному разрыву с 5′-конца повреждения. Образование обоих разрывов является ATP-зависимым, и их расположение на ДНК высокоспецифично. Как правило, происходят разрывы 5-й и 24-й фосфодиэфирных связей соответственно от 3′- и 5′-концов поврежденных участков. Однако расположение точек разрывов может варьироваться.
Таким образом, в результате подобных одноцепочечных надрезов ДНК может освобождаться фрагмент длиной 24-32 нуклеотида с преобладанием фрагментов длиной 27-29 нуклеотидов. На расположение сайтов одноцепочечных разрывов влияют характер повреждения и последовательности нуклеотидов (контекст), окружающих поврежденный участок. Ту же самую картину инцизии обнаруживают in vivo в ооцитах Xenopus и у Schizosaccharomyces pombe. На этом основании делают вывод об универсальном механизме эксцизионной репарации у эукариот.
Репаративный синтез ДНК у человека является PCNA-зависимым,
т.е. может осуществляться с участием ДНК-полимераз Polдельта и Polэпсилон. PCNA связывается с системой праймер-матрица под действием фактора репликации RFC, откуда следует, что последний также участвует в репаративном синтезе ДНК. В опытах с бесклеточными системами моноклональные антитела к Polдельта специфически подавляют репаративный синтез. Однако оказалось, что в тех же высокоочищенных бесклеточных системах вместо Polдельта с аналогичным эффектом могут быть использованы Polэпсилон и даже фрагмент Кленова ДНК-полимеразы I E. coli. Это означает, что реконструированные из очищенных компонентов бесклеточные системы лишь в ограниченной степени имитируют биохимические процессы, происходящие в живых клетках. Считается, что обе ДНК-полимеразы — Polдельта и Polэпсилон участвуют в репаративном синтезе ДНК у человека.
ПАРП: модели управления процессом эксцизионной репарации ДНК
ПАРП — один из первых ядерных факторов, распознающих повреждение ДНК , и поэтому в идеальном случае управляет запуском механизма
репарации ДНК в живых клетках с места повреждения ДНК. Эта модель поддерживается идентификацией ЭР -комплекса, включающего ПАРП , XRCC1 , ДНК-лигазу III и ДНК-полимеразу бета . Присутствие ПАРП в таком мультипротеиновом комплексе доказывает, что этот фермент может направлять аппарат репарации ДНК к сайтам повреждения ДНК in vivo и облегчает осуществление репарации по этому пути.
XRCC1-белок действует как молекулярные «леса», формируя ЭР-комплекс путем индивидуального взаимодействия с каждым компонентом. С тех пор, как была обнаружена возможность поли(АДФ-рибозил)ирования XRCC1-белка in vitro, можно предположить, что ПАРП способен регулировать активность комплекса путем модифицирования XRCC1-белка in vivo и нарушать его способность взаимодействовать с другими компонентами комплекса. Было показано, что сверхэкспрессия XRCC1-белка подавляет активность ПАРП в живых клетках.
Аналогично ДНК-лигаза III ингибирует активность ПАРП in vitro, когда ее количества превышают количества ПАРП.
ПАРП может также рекрутировать факторы репарации ДНК путем модификации хроматиновых белков. Длинные цепи ПАР действительно способны направить ферменты репарации к сайтам разрывов ДНК значительно быстрее, нежели если они ищут повреждение сами по всему ядру. Такая модель согласуется с активностью ПАРП перед и/или после удаления поврежденных оснований.
NER: структура и функции белков
В табл. I.21 суммированы свойства белков животных, участвующих в NER. Большинство этих белков существует in vivo в виде комплексов, поэтому ферментативные активности, обнаруживаемые у отдельных белков в очищенном состоянии, могут не иметь прямого отношения к их функциям в системе NER.
XPA — белок с молекулярной массой 31 кДа, обладает доменом типа «цинковые пальцы», участвует в распознавании поврежденного участка ДНК, взаимодействует с другими компонентами системы и может функционировать в качестве фактора нуклеации для экзонуклеазы. XPA взаимодействует своим N-концевым доменом
с гетеродимером ERCC1-XPF, а С-концевым доменом — с TFIIH.
Белок RPA (HSSB) образует комплекс с XPA и усиливает его специфичность в отношении поврежденной ДНК. RPA (HSSB) — тример, состоящий из белковых субъединиц р70, р34 и р11, необходим для репликации ДНК и репаративного синтеза, а также для прохождения этапа двойного надреза ДНК во время эксцизионной репарации. Он обладает умеренным сродством к поврежденной ДНК.
TFIIH — олигомерный комплекс, в состав которого входят белки р89, р80, р62, р44, р41, р38 и р34. Этот белковый комплекс был открыт как один из семи основных факторов транскрипции, необходимых для эффективного функционирования РНК-полимеразы II . Субъединица р89 идентична белку репаративного комплекса XPB. Обнаружено отсутствие функциональной комплементации между бесклеточными экстрактами клеток с мутантными белками XPB и XPD, определяемой по восстановлению репарирующей активности в смешанных экстрактах. Комплекс TFIIH представляет собой
фактор репаративной системы. Белки XPB и XPD являются ДНК-зависимыми АТРазами, обладают хеликазными доменами и могут (как и сам фактор TFIIH) вызывать диссоциацию гибридов, образованных между короткими фрагментами ДНК и одноцепочечной ДНК.
XPC — белок с молекулярной массой около 125 кДа, существует в виде гетеродимера в комплексе с белком р58, который является гомологом белка Rad23 дрожжей (HHR23B). XPC слабо связывается с TFIIH и очень прочно — с одноцепочечной ДНК.
ERCC1/XPF — прочный белковый комплекс, с которым взаимодействует белок XPA, обладающий эндонуклеазной активностью, специфичной в отношении одноцепочечной ДНК.
XPG — белковый комплекс, обладающий эндонуклеазной активностью, специфичной в отношении одноцепочечной ДНК; вовлекается в эксцизионный комплекс посредством взаимодействия с TFIIH и RPA.
ЭРН (NER): генетика
Гены NER E. coli, uvrA, uvrB и uvrC не обнаруживают гомологии с соответствующими генами человека. Гены NER дрожжей и человека
высокогомологичны, и энзимология эксцизионной репарации также обладает большим сходством. По крайней мере, три заболевания у человека вызываются генетическими нарушениями системы эксцизионной репарации: пигментная ксеродерма, синдром Кокейна и трихотиодистрофия.
Кожа больных пигментной ксеродермой обладает повышенной чувствительностью к дневному свету, что проявляется в виде фотодерматозов, включая рак кожи. В ряде случаев отмечены аномалии нервной системы, причиной которых являются мутации в одном из семи генов: XPA, XPB, XPC, XPD, XPE, XPF, XPG. Однако описаны больные с классическими симптомами пигментной ксеродермы, но с ненарушенной системой NER. Для клеток этих больных характерны изменения в так называемой пострепликативной репарации .
Больным с синдромом Кокейна присущи нарушения роста, умственная отсталость, катаракты, повышенная чувствительность к свету с сопутствующими дерматозами. Обнаружены мутации в двух группах генов, приводящие к этому заболеванию. У больных
с мутантными генами CS-A или CS-B клетки способны нормально репарировать УФ-повреждения ДНК. У другой группы больных обнаружены мутации в генах XPB, XPD или XPG.
У больных трихотиодистрофией со смешанными симптомами выявлены мутации в генах XPB или XPD. Классические симптомы этого заболевания, по-видимому, являются следствием мутации в гене транскрипционного фактора TFIIH.
Получение мутантов с измененной NER у грызунов позволило разбить такие гены на 11 групп комплементации, большинство из которых соответствует группам комплементации XP и CS человека. Часть соответствующих генов человека удалось клонировать, используя их способность исправлять (комплементировать) генетические дефекты в культивируемых мутантных клетках грызунов. Эти гены получили название кросс-комплементирующих генов эксцизионной репарации ( ERCC — excision repair cross complementing ). Среди них гены XPE и ERCC6-ERCC11 не требовались для прохождения основных реакций эксцизионной репарации, и их функция неизвестна.
Эксцизионная репарация нуклеотидов (ЭРН) E. coli
У E. coli эксцинуклеаза формируется, в результате димеризации двух молекул белка UvrА в присутствии АТР и связывания с одной молекулой UvrB. Гетеротример UvrA2B связывается с ДНК и с помощью 5′-3′-ДНК-геликазной активности перемещается вдоль ДНК в поисках повреждения. Такова предполагаемая картина узнавания эксцинуклеазой UvrAВС неспецифического повреждения в ДНК [ Hoeijmakers J., 1993 ]. Результатом узнавания повреждения этим комплексом явится внедрение субъединицы UvrB в ДНК, сопровождающееся конформационными изменениями ДНК в сайте внедрения ( изломы , локальная денатурация), диссоциация обеих субъединиц UvrА из комплекса и последующее связывание с ним молекул UvrD и UvrC. Гетеродимер UvrBC из состава нового комплекса делает два разрыва в поврежденной нити ДНК на расстоянии 8 н. с 5′-конца от повреждения и 5 н. с 3′-конца, катализируемых субъединицами UvrC и UvrB соответственно, что приводит к появлению 12-13-мерного олигонуклеотида,
вытесняемого из комплекса с помощью геликазы UvrD. Образующаяся при этом брешь заполняется ДНК-полимеразой I, синтезирующей ДНК по неповрежденной матрице, и сшивается ДНК-лигазой. Описанный процесс зависит от АТР. Действительно, АТР стимулирует димеризацию молекул UvrA, гидролиз АТР необходим для образования комплекса UvrA2B и проявления его геликазной активности, АТР необходим для формирования комплекса UvrB-ДНК и, наконец, для описанной выше бимодальной инцизии ДНК [ Friedberg E.C., Walker G.C., ea., 1991 ]. У E. coli транскрипционно-зависимую ветвь ЭРН (ТЭРН) осуществляет продукт гена mfd (mutation frequency decline). Этот ген был открыт за 35 лет до того, как его продукт признали фактором TRCF. Последний способствует диссоциации РНК-полимеразы от дефектной транскрибируемой нити ДНК, и связывается с субъединицей UvrA эксцинуклеазного комплекса. Иными словами, именно белок Mfd отыскивает повреждения на транскрибируемой нити и направляет ТЭРН на эту мишень.
Согласно недавним наблюдениям, два главных белка системы ДКНО — MutL и MutS — необходимы для ТЭРН [106 Melon I., Champl G.N., 1996106]. Причина такой взаимосвязи систем ДКНО и ТЭРН или заимствования белков MutL и MutS для выполнения сходных функций пока остается загадочной, хотя сам факт несомненен и нашел подтверждение также и для белков Msh2, Mlh1, Pms1 и Msh3 у S. cerevisiae [ Sweder K.S., ea, 1996 ].
Эксцизионная репарация нуклеотидов (ЭРН) у человека
Большому прогрессу в раскрытии механизма ЭРН у эукариот мы обязаны наследственному заболеванию человека — пигментной ксеродерме .
Изучение молекулярных основ этого заболевания выявили их сопряженность с дефектами системы ЭРН, результатом чего и явилось раскрытие генетического контроля ЭРН. Комплементационный анализ различных клеточных линий ХР определил 8 комплементационных групп (7 от XPA до XPG и одна «вариантная форма» XPV) [ Friedberg E.C., Walker G.C., ea., 1991 ]), а комплементационная
коррекция возможных мутантных линий клеток китайского хомячка, чувствительных к УФ-свету, способствовала выявлению дополнительных генов системы ЭРН. Последние получили название гены ERCC (excision repair cross-complimenting), и поэтому в названии генов и белков ЭРН используются обе аббревиатуры. Хотя детали процесса ЭРН у человека не ясны, так как не определена роль целого ряда генов, в первом приближении он выглядит так ( табл. 3 ) [ Lehmann A.R., 1995 , Lindahl T., ea, 1997 , Hoeijmakers J., 1993 ]. Белок ХРА в комплексе с онДНК-связующим белком RPA ( репликационным белком А ), транслоцируясь вдоль онДНК, опознает конформационнoе повреждениe. Взаимодействуя через другой участок белка ХРА с базальным фактором транскрипции TFIIH (две из субъединиц которого, белки ХРВ и ХРD , обладают геликазной активностью с противоположной ориентацией раскручивания днДНК), они образуют комплекс, который расплетает ДНК вокруг повреждения (в состав этого комплекса входит и белок ХРС
с неясными функциями). Через третий участок белка ХРА к комплексу примыкает гетеродимер ERCC1-XPF , который вносит однонитевой разрыв в ДНК с 5′-конца на расстоянии 16-25 н. от повреждения, тогда как белок XPG , входящий в комплекс белков эксцинуклеазы через взаимодействие с белком RPA, делает надрез с 3′-конца на расстоянии 2-9 н. (в разрезании принимает участие и белок ХРС , а белок ХРЕ активирует реакцию). В результате бимодальной инцизии участок ДНК размером около 29 н. высвобождается, а образующаяся брешь ресинтезируется с помощью ДНК-полимеразы епсилон или ДНК-полимеразы дельта , сопутствующего репликации фактора PCNА , репликационного фактора C-RFС и ДНК-лигазы I .
Представленная картина во многом основана на реконструировании процесса в открытой системе с участием 10 хорошо очищенных белков [ Wood R.D., 1994 ]. Как видно, эукариотическая система ЭРН лишь функционально подобна прокариотической. В ней задействовано значительно больше белков, число которых должно еще возрасти
за счет белков, осуществляющих разборку и сборку хроматина. Раскрытие природы ТЭРН у человека также связывают с пониманием молекулярного дефекта,приводящего к развитию двух мультисистемных генетических заболеваний — синдрома Кокейна (СS) и трихотиодистрофии ( ТТD ). При обоих заболеваниях соматические клетки больных оказались неспособны к ТЭРН. Классические случаи CS оказались связанными с повреждениями в двух генах, CSA и CSB, а редкие смешанные формы CS+XP (CS с дополнительными симптомами ХР), с повреждениями в генах ХРВ, ХРD и ХРG. При ТТD дефекты были обнаружены также в генах XPB и XPD и в новом пока не клонированном гене ТТDА, продукт которого является частью корового домена транскрипционного фактора TFIIH [ Lehmann A.R., 1995 , Lindahl T., ea, 1997 ].
Фактор TFIIH состоит из 6 субъединиц, две из которых — белки XPB и XPD. Связываясь с белками РНК-полимеразного комплекса, кор TFIIH принимает участие в инициировании транскрипции [ Buratowski S., 1994 ], a вкупе с белками репарационного
комплекса кор этого фактора участвует в ЭРН. Если допустить, что белки CSA и CSB способствуют превращению транскрипционного комплекса в репарационнный [ Lindahl T., ea, 1997 , Bregman D.B., 1996 ], то следствием повреждения этих белков может стать дефектность в ТЭРН. Таким образом, либо прямыми воздействиями на коровый домен фактора TFIIH (мутации в генах XPB, XPD и TTDA), либо косвенными (через гены CSA и CSB) можно модифицировать способность этого фактора к диссоциации из РНК- полимеразного комплекса для участия в ТЭРН. Что касается белка XPG, то он, подобно своему гомологу из S. cereivisiae — белку Rad2 [ Bregman D.B., 1996 ], способен взаимодействовать c TFIIH, что допускает возможность его воздействия на TFIIH для участия в ТЭРН. Таковы гипотетические объяснения взаимосвязи молекулярных дефектов при CS и ТTD с нарушениями ТЭРН, которые могут быть полезными для раскрытия механизма ТЭРН у эукариот. Связь же между молекулярными дефектами и клиническими проявлениями
рассматриваемых наследственных заболеваний пока не имеет даже упрощенных толкований [ Kolodner R., 1995 ].
Так случилось, что изучение молекулярных механизмов ЭРН и ТЭРН на клетках человека развивались параллельно, или даже опережая исследования на популярной эукариотической модели дрожжей S. cerevisiae. В табл. 3 представлены известные функциональные аналоги системы ЭРН у дрожжей. Несомненно, однако, что при исследовании детального механизма ЭРН именно эта модельная система будет играть ведущую роль. Например, немаловажной особенностью системы ЭРН у E. coli является ее связь с SOS-функциями клетки. Действительно, центральные гены системы ЭРН, uvrA и uvrB, находятся под контролем SOS-регулона [ Friedberg E.C., Walker G.C., ea., 1991 ]. Системы, подобной SOS, у эукариот пока не обнаружено. Однако целый ряд генов системы ЭРН у дрожжей, таких, как RAD2, RAD7, RAD23, CDC8 и CDC9, индуцируется при повреждении ДНК, а последние два гена дополнительно регулируются клеточным циклом [ Friedberg
E.C., Walker G.C., ea., 1991 ].
Природу этой индукции еще предстоит выяснить.
Макеты страниц
Было установлено, что частота ошибок при репликации ДНК Е. coli не превышает 1 на  нуклеотидов. Поскольку хромосома Е. coli содержит приблизительно
нуклеотидов. Поскольку хромосома Е. coli содержит приблизительно  пар оснований, на 10000 клеток, претерпевших один цикл деления, встраивается всего один неправильный нуклеотид.
пар оснований, на 10000 клеток, претерпевших один цикл деления, встраивается всего один неправильный нуклеотид.
Долгое время считалось, что столь высокая степень точности воспроизведения генетической информации целиком определяется точностью уотсон-криковского спаривания между матричной и новообразованной (дочерней) цепями, однако в результате последующего анализа выяснилось, что если бы точность репликации зависела исключительно от правильности спаривания оснований, то частота ошибок была бы значительно выше — приблизительно 1 на 104-105 остатков. Следовательно, чтобы объяснить такую низкую частоту ошибок при репликации in vivo, необходимо предположить участие в процессе репликации еще какого-то одного или нескольких факторов.
Более детальное изучение свойств высокоочищенных ДНК-полимераз позволило получить по крайней мере частичный ответ на вопрос о природе этих факторов. Напомним, что ДНК-полимеразы I и III обладают тремя различными ферментативными активностями. Мы уже видели, как фермент функционирует в качестве полимеразы, а также как он может удалять нуклеотидные остатки с 5-конца фрагмента ДНК. Однако 3-экзонуклеазная активность ДНК-полимераз I и III очень озадачивала исследователей, ибо она означала, что эти ферменты способны «пятиться», отщепляя З-концевые нуклеотиды в направлении, противоположном тому, в котором они действуют как полимеразы. З-экзону-клеазная активность ДНК-полимераз I и III — это средство проверки новосинтезированной цепи ДНК и исправления ошибок, сделанных ферментом при его работе в качестве полимеразы. Если ДНК-полимераза встраивает неправильный нуклеотид, то фермент сам может распознать неспособность этого нуклеотида образовать правильную пару с соответствующим нуклеотидом матрицы (рис. 28-15). В этом случае фермент возвращается назад и отщепляет неправильный нуклеотид с З-конца цепи, после чего полимераза продолжает присоединять правильные нуклеотиды, т.е. возобновляет свое обычное продвижение в направлении 

Рис. 28-15. Исправление ошибок с помощью 3-зкзонуклеазной активности ДНК-полимеразы.
Таким образом, по мере перемещения репликативной вилки вдоль матрицы осуществляется проверка каждого встроенного нуклеотида. Корректирующее действие ДНК-полимеразы очень эффективно; благодаря ему точность репликации повышается как минимум в 104 раз. Суммарная ошибка возникает в результате ошибок, допускаемых ферментом в ходе полимеризации и в процессе исправления их при корректировке; она не превышает одной ошибки на  нуклеотидных остатков.
нуклеотидных остатков.
Очень важно отметить, что процесс репликации протекает со значительно более высокой степенью точности, чем процессы транскрипции и трансляции. Частые ошибки в репликации подвергли бы большому риску сохранность видов и их жизнеспособность.
Ошибки же в транскрипции и трансляции гораздо менее опасны, поскольку они влияют на образование РНК или белка только в одной клетке и не изменяют всю последующую родословную вида. Корректировка с помощью ДНК-полимеразы — это, вероятно, лишь один из путей, обеспечивающих высокую точность репликации. Возможно, исключительно сложная организация репликативного процесса и участие в нем множества белков необходимы для достижения именно этой цели. Интересно, что некоторые эукариотические ДНК-полимеразы не осуществляют корректировку. По-видимому, эукариоты обеспечивают точность и надежность процесса репликации с помощью каких-то других средств.

DNA damage resulting in multiple broken chromosomes
DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome.[1] In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day.[2] Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell’s ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell’s genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur, including double-strand breaks and DNA crosslinkages (interstrand crosslinks or ICLs).[3][4] This can eventually lead to malignant tumors, or cancer as per the two hit hypothesis.
The rate of DNA repair is dependent on many factors, including the cell type, the age of the cell, and the extracellular environment. A cell that has accumulated a large amount of DNA damage, or one that no longer effectively repairs damage incurred to its DNA, can enter one of three possible states:
- an irreversible state of dormancy, known as senescence
- cell suicide, also known as apoptosis or programmed cell death
- unregulated cell division, which can lead to the formation of a tumor that is cancerous
The DNA repair ability of a cell is vital to the integrity of its genome and thus to the normal functionality of that organism. Many genes that were initially shown to influence life span have turned out to be involved in DNA damage repair and protection.[5]
Paul Modrich talks about himself and his work in DNA repair.
The 2015 Nobel Prize in Chemistry was awarded to Tomas Lindahl, Paul Modrich, and Aziz Sancar for their work on the molecular mechanisms of DNA repair processes.[6][7]
DNA damage[edit]
DNA damage, due to environmental factors and normal metabolic processes inside the cell, occurs at a rate of 10,000 to 1,000,000 molecular lesions per cell per day.[2] While this constitutes only 0.000165% of the human genome’s approximately 6 billion bases, unrepaired lesions in critical genes (such as tumor suppressor genes) can impede a cell’s ability to carry out its function and appreciably increase the likelihood of tumor formation and contribute to tumour heterogeneity.
The vast majority of DNA damage affects the primary structure of the double helix; that is, the bases themselves are chemically modified. These modifications can in turn disrupt the molecules’ regular helical structure by introducing non-native chemical bonds or bulky adducts that do not fit in the standard double helix. Unlike proteins and RNA, DNA usually lacks tertiary structure and therefore damage or disturbance does not occur at that level. DNA is, however, supercoiled and wound around «packaging» proteins called histones (in eukaryotes), and both superstructures are vulnerable to the effects of DNA damage.
Sources[edit]
DNA damage can be subdivided into two main types:
- endogenous damage such as attack by reactive oxygen species produced from normal metabolic byproducts (spontaneous mutation), especially the process of oxidative deamination
- also includes replication errors
- exogenous damage caused by external agents such as
- ultraviolet [UV 200–400 nm] radiation from the sun or other artificial light sources
- other radiation frequencies, including x-rays and gamma rays
- hydrolysis or thermal disruption
- certain plant toxins
- human-made mutagenic chemicals, especially aromatic compounds that act as DNA intercalating agents
- viruses[8]
The replication of damaged DNA before cell division can lead to the incorporation of wrong bases opposite damaged ones. Daughter cells that inherit these wrong bases carry mutations from which the original DNA sequence is unrecoverable (except in the rare case of a back mutation, for example, through gene conversion).
Types[edit]
There are several types of damage to DNA due to endogenous cellular processes:
- oxidation of bases [e.g. 8-oxo-7,8-dihydroguanine (8-oxoG)] and generation of DNA strand interruptions from reactive oxygen species,
- alkylation of bases (usually methylation), such as formation of 7-methylguanosine, 1-methyladenine, 6-O-Methylguanine
- hydrolysis of bases, such as deamination, depurination, and depyrimidination.
- «bulky adduct formation» (e.g., benzo[a]pyrene diol epoxide-dG adduct, aristolactam I-dA adduct)
- mismatch of bases, due to errors in DNA replication, in which the wrong DNA base is stitched into place in a newly forming DNA strand, or a DNA base is skipped over or mistakenly inserted.
- Monoadduct damage cause by change in single nitrogenous base of DNA
- Diadduct damage
Damage caused by exogenous agents comes in many forms. Some examples are:
- UV-B light causes crosslinking between adjacent cytosine and thymine bases creating pyrimidine dimers. This is called direct DNA damage.
- UV-A light creates mostly free radicals. The damage caused by free radicals is called indirect DNA damage.
- Ionizing radiation such as that created by radioactive decay or in cosmic rays causes breaks in DNA strands. Intermediate-level ionizing radiation may induce irreparable DNA damage (leading to replicational and transcriptional errors needed for neoplasia or may trigger viral interactions) leading to pre-mature aging and cancer.
- Thermal disruption at elevated temperature increases the rate of depurination (loss of purine bases from the DNA backbone) and single-strand breaks. For example, hydrolytic depurination is seen in the thermophilic bacteria, which grow in hot springs at 40–80 °C.[9][10] The rate of depurination (300 purine residues per genome per generation) is too high in these species to be repaired by normal repair machinery, hence a possibility of an adaptive response cannot be ruled out.
- Industrial chemicals such as vinyl chloride and hydrogen peroxide, and environmental chemicals such as polycyclic aromatic hydrocarbons found in smoke, soot and tar create a huge diversity of DNA adducts- ethenobases, oxidized bases, alkylated phosphotriesters and crosslinking of DNA, just to name a few.
UV damage, alkylation/methylation, X-ray damage and oxidative damage are examples of induced damage. Spontaneous damage can include the loss of a base, deamination, sugar ring puckering and tautomeric shift. Constitutive (spontaneous) DNA damage caused by endogenous oxidants can be detected as a low level of histone H2AX phosphorylation in untreated cells.[11]
Nuclear versus mitochondrial[edit]
In human cells, and eukaryotic cells in general, DNA is found in two cellular locations – inside the nucleus and inside the mitochondria. Nuclear DNA (nDNA) exists as chromatin during non-replicative stages of the cell cycle and is condensed into aggregate structures known as chromosomes during cell division. In either state the DNA is highly compacted and wound up around bead-like proteins called histones. Whenever a cell needs to express the genetic information encoded in its nDNA the required chromosomal region is unravelled, genes located therein are expressed, and then the region is condensed back to its resting conformation. Mitochondrial DNA (mtDNA) is located inside mitochondria organelles, exists in multiple copies, and is also tightly associated with a number of proteins to form a complex known as the nucleoid. Inside mitochondria, reactive oxygen species (ROS), or free radicals, byproducts of the constant production of adenosine triphosphate (ATP) via oxidative phosphorylation, create a highly oxidative environment that is known to damage mtDNA. A critical enzyme in counteracting the toxicity of these species is superoxide dismutase, which is present in both the mitochondria and cytoplasm of eukaryotic cells.
Senescence and apoptosis[edit]
Senescence, an irreversible process in which the cell no longer divides, is a protective response to the shortening of the chromosome ends, called telomeres. The telomeres are long regions of repetitive noncoding DNA that cap chromosomes and undergo partial degradation each time a cell undergoes division (see Hayflick limit).[12] In contrast, quiescence is a reversible state of cellular dormancy that is unrelated to genome damage (see cell cycle). Senescence in cells may serve as a functional alternative to apoptosis in cases where the physical presence of a cell for spatial reasons is required by the organism,[13] which serves as a «last resort» mechanism to prevent a cell with damaged DNA from replicating inappropriately in the absence of pro-growth cellular signaling. Unregulated cell division can lead to the formation of a tumor (see cancer), which is potentially lethal to an organism. Therefore, the induction of senescence and apoptosis is considered to be part of a strategy of protection against cancer.[14]
Mutation[edit]
It is important to distinguish between DNA damage and mutation, the two major types of error in DNA. DNA damage and mutation are fundamentally different. Damage results in physical abnormalities in the DNA, such as single- and double-strand breaks, 8-hydroxydeoxyguanosine residues, and polycyclic aromatic hydrocarbon adducts. DNA damage can be recognized by enzymes, and thus can be correctly repaired if redundant information, such as the undamaged sequence in the complementary DNA strand or in a homologous chromosome, is available for copying. If a cell retains DNA damage, transcription of a gene can be prevented, and thus translation into a protein will also be blocked. Replication may also be blocked or the cell may die.
In contrast to DNA damage, a mutation is a change in the base sequence of the DNA. A mutation cannot be recognized by enzymes once the base change is present in both DNA strands, and thus a mutation cannot be repaired. At the cellular level, mutations can cause alterations in protein function and regulation. Mutations are replicated when the cell replicates. In a population of cells, mutant cells will increase or decrease in frequency according to the effects of the mutation on the ability of the cell to survive and reproduce.
Although distinctly different from each other, DNA damage and mutation are related because DNA damage often causes errors of DNA synthesis during replication or repair; these errors are a major source of mutation.
Given these properties of DNA damage and mutation, it can be seen that DNA damage is a special problem in non-dividing or slowly-dividing cells, where unrepaired damage will tend to accumulate over time. On the other hand, in rapidly dividing cells, unrepaired DNA damage that does not kill the cell by blocking replication will tend to cause replication errors and thus mutation. The great majority of mutations that are not neutral in their effect are deleterious to a cell’s survival. Thus, in a population of cells composing a tissue with replicating cells, mutant cells will tend to be lost. However, infrequent mutations that provide a survival advantage will tend to clonally expand at the expense of neighboring cells in the tissue. This advantage to the cell is disadvantageous to the whole organism because such mutant cells can give rise to cancer. Thus, DNA damage in frequently dividing cells, because it gives rise to mutations, is a prominent cause of cancer. In contrast, DNA damage in infrequently-dividing cells is likely a prominent cause of aging.[15]
Mechanisms[edit]
Cells cannot function if DNA damage corrupts the integrity and accessibility of essential information in the genome (but cells remain superficially functional when non-essential genes are missing or damaged). Depending on the type of damage inflicted on the DNA’s double helical structure, a variety of repair strategies have evolved to restore lost information. If possible, cells use the unmodified complementary strand of the DNA or the sister chromatid as a template to recover the original information. Without access to a template, cells use an error-prone recovery mechanism known as translesion synthesis as a last resort.
Damage to DNA alters the spatial configuration of the helix, and such alterations can be detected by the cell. Once damage is localized, specific DNA repair molecules bind at or near the site of damage, inducing other molecules to bind and form a complex that enables the actual repair to take place.
Direct reversal[edit]
Cells are known to eliminate three types of damage to their DNA by chemically reversing it. These mechanisms do not require a template, since the types of damage they counteract can occur in only one of the four bases. Such direct reversal mechanisms are specific to the type of damage incurred and do not involve breakage of the phosphodiester backbone. The formation of pyrimidine dimers upon irradiation with UV light results in an abnormal covalent bond between adjacent pyrimidine bases. The photoreactivation process directly reverses this damage by the action of the enzyme photolyase, whose activation is obligately dependent on energy absorbed from blue/UV light (300–500 nm wavelength) to promote catalysis.[16] Photolyase, an old enzyme present in bacteria, fungi, and most animals no longer functions in humans,[17] who instead use nucleotide excision repair to repair damage from UV irradiation. Another type of damage, methylation of guanine bases, is directly reversed by the enzyme methyl guanine methyl transferase (MGMT), the bacterial equivalent of which is called ogt. This is an expensive process because each MGMT molecule can be used only once; that is, the reaction is stoichiometric rather than catalytic.[18] A generalized response to methylating agents in bacteria is known as the adaptive response and confers a level of resistance to alkylating agents upon sustained exposure by upregulation of alkylation repair enzymes.[19] The third type of DNA damage reversed by cells is certain methylation of the bases cytosine and adenine.
Single-strand damage[edit]

Structure of the base-excision repair enzyme uracil-DNA glycosylase excising a hydrolytically-produced uracil residue from DNA. The uracil residue is shown in yellow.
When only one of the two strands of a double helix has a defect, the other strand can be used as a template to guide the correction of the damaged strand. In order to repair damage to one of the two paired molecules of DNA, there exist a number of excision repair mechanisms that remove the damaged nucleotide and replace it with an undamaged nucleotide complementary to that found in the undamaged DNA strand.[18]
- Base excision repair (BER): damaged single bases or nucleotides are most commonly repaired by removing the base or the nucleotide involved and then inserting the correct base or nucleotide. In base excision repair, a glycosylase[20] enzyme removes the damaged base from the DNA by cleaving the bond between the base and the deoxyribose. These enzymes remove a single base to create an apurinic or apyrimidinic site (AP site).[20] Enzymes called AP endonucleases nick the damaged DNA backbone at the AP site. DNA polymerase then removes the damaged region using its 5’ to 3’ exonuclease activity and correctly synthesizes the new strand using the complementary strand as a template.[20] The gap is then sealed by enzyme DNA ligase.[21]
- Nucleotide excision repair (NER): bulky, helix-distorting damage, such as pyrimidine dimerization caused by UV light is usually repaired by a three-step process. First the damage is recognized, then 12-24 nucleotide-long strands of DNA are removed both upstream and downstream of the damage site by endonucleases, and the removed DNA region is then resynthesized.[22] NER is a highly evolutionarily conserved repair mechanism and is used in nearly all eukaryotic and prokaryotic cells.[22] In prokaryotes, NER is mediated by Uvr proteins.[22] In eukaryotes, many more proteins are involved, although the general strategy is the same.[22]
- Mismatch repair systems are present in essentially all cells to correct errors that are not corrected by proofreading. These systems consist of at least two proteins. One detects the mismatch, and the other recruits an endonuclease that cleaves the newly synthesized DNA strand close to the region of damage. In E. coli , the proteins involved are the Mut class proteins: MutS, MutL, and MutH. In most Eukaryotes, the analog for MutS is MSH and the analog for MutL is MLH. MutH is only present in bacteria. This is followed by removal of damaged region by an exonuclease, resynthesis by DNA polymerase, and nick sealing by DNA ligase.[23]
Double-strand breaks[edit]
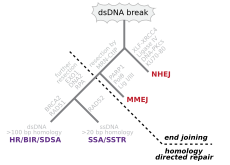
The main double-strand break repair pathways
Double-strand breaks, in which both strands in the double helix are severed, are particularly hazardous to the cell because they can lead to genome rearrangements. In fact, when a double-strand break is accompanied by a cross-linkage joining the two strands at the same point, neither strand can be used as a template for the repair mechanisms, so that the cell will not be able to complete mitosis when it next divides, and will either die or, in rare cases, undergo a mutation.[3][4] Three mechanisms exist to repair double-strand breaks (DSBs): non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homologous recombination (HR):[18][24]

DNA ligase, shown above repairing chromosomal damage, is an enzyme that joins broken nucleotides together by catalyzing the formation of an internucleotide ester bond between the phosphate backbone and the deoxyribose nucleotides.
- In NHEJ, DNA Ligase IV, a specialized DNA ligase that forms a complex with the cofactor XRCC4, directly joins the two ends.[25] To guide accurate repair, NHEJ relies on short homologous sequences called microhomologies present on the single-stranded tails of the DNA ends to be joined. If these overhangs are compatible, repair is usually accurate.[26][27][28][29] NHEJ can also introduce mutations during repair. Loss of damaged nucleotides at the break site can lead to deletions, and joining of nonmatching termini forms insertions or translocations. NHEJ is especially important before the cell has replicated its DNA, since there is no template available for repair by homologous recombination. There are «backup» NHEJ pathways in higher eukaryotes.[30] Besides its role as a genome caretaker, NHEJ is required for joining hairpin-capped double-strand breaks induced during V(D)J recombination, the process that generates diversity in B-cell and T-cell receptors in the vertebrate immune system.[31]
- MMEJ starts with short-range end resection by MRE11 nuclease on either side of a double-strand break to reveal microhomology regions.[32] In further steps,[33] Poly (ADP-ribose) polymerase 1 (PARP1) is required and may be an early step in MMEJ. There is pairing of microhomology regions followed by recruitment of flap structure-specific endonuclease 1 (FEN1) to remove overhanging flaps. This is followed by recruitment of XRCC1–LIG3 to the site for ligating the DNA ends, leading to an intact DNA. MMEJ is always accompanied by a deletion, so that MMEJ is a mutagenic pathway for DNA repair.[34]
- HR requires the presence of an identical or nearly identical sequence to be used as a template for repair of the break. The enzymatic machinery responsible for this repair process is nearly identical to the machinery responsible for chromosomal crossover during meiosis. This pathway allows a damaged chromosome to be repaired using a sister chromatid (available in G2 after DNA replication) or a homologous chromosome as a template. DSBs caused by the replication machinery attempting to synthesize across a single-strand break or unrepaired lesion cause collapse of the replication fork and are typically repaired by recombination.
In an in vitro system, MMEJ occurred in mammalian cells at the levels of 10–20% of HR when both HR and NHEJ mechanisms were also available.[32]
The extremophile Deinococcus radiodurans has a remarkable ability to survive DNA damage from ionizing radiation and other sources. At least two copies of the genome, with random DNA breaks, can form DNA fragments through annealing. Partially overlapping fragments are then used for synthesis of homologous regions through a moving D-loop that can continue extension until complementary partner strands are found. In the final step, there is crossover by means of RecA-dependent homologous recombination.[35]
Topoisomerases introduce both single- and double-strand breaks in the course of changing the DNA’s state of supercoiling, which is especially common in regions near an open replication fork. Such breaks are not considered DNA damage because they are a natural intermediate in the topoisomerase biochemical mechanism and are immediately repaired by the enzymes that created them.
Another type of DNA double-strand breaks originates from the DNA heat-sensitive or heat-labile sites. These DNA sites are not initial DSBs. However, they convert to DSB after treating with elevated temperature. Ionizing irradiation can induces a highly complex form of DNA damage as clustered damage. It consists of different types of DNA lesions in various locations of the DNA helix. Some of these closely located lesions can probably convert to DSB by exposure to high temperatures. But the exact nature of these lesions and their interactions is not yet known[36]
Translesion synthesis[edit]
Translesion synthesis (TLS) is a DNA damage tolerance process that allows the DNA replication machinery to replicate past DNA lesions such as thymine dimers or AP sites.[37] It involves switching out regular DNA polymerases for specialized translesion polymerases (i.e. DNA polymerase IV or V, from the Y Polymerase family), often with larger active sites that can facilitate the insertion of bases opposite damaged nucleotides. The polymerase switching is thought to be mediated by, among other factors, the post-translational modification of the replication processivity factor PCNA. Translesion synthesis polymerases often have low fidelity (high propensity to insert wrong bases) on undamaged templates relative to regular polymerases. However, many are extremely efficient at inserting correct bases opposite specific types of damage. For example, Pol η mediates error-free bypass of lesions induced by UV irradiation, whereas Pol ι introduces mutations at these sites. Pol η is known to add the first adenine across the T^T photodimer using Watson-Crick base pairing and the second adenine will be added in its syn conformation using Hoogsteen base pairing. From a cellular perspective, risking the introduction of point mutations during translesion synthesis may be preferable to resorting to more drastic mechanisms of DNA repair, which may cause gross chromosomal aberrations or cell death. In short, the process involves specialized polymerases either bypassing or repairing lesions at locations of stalled DNA replication. For example, Human DNA polymerase eta can bypass complex DNA lesions like guanine-thymine intra-strand crosslink, G[8,5-Me]T, although it can cause targeted and semi-targeted mutations.[38] Paromita Raychaudhury and Ashis Basu[39] studied the toxicity and mutagenesis of the same lesion in Escherichia coli by replicating a G[8,5-Me]T-modified plasmid in E. coli with specific DNA polymerase knockouts. Viability was very low in a strain lacking pol II, pol IV, and pol V, the three SOS-inducible DNA polymerases, indicating that translesion synthesis is conducted primarily by these specialized DNA polymerases.
A bypass platform is provided to these polymerases by Proliferating cell nuclear antigen (PCNA). Under normal circumstances, PCNA bound to polymerases replicates the DNA. At a site of lesion, PCNA is ubiquitinated, or modified, by the RAD6/RAD18 proteins to provide a platform for the specialized polymerases to bypass the lesion and resume DNA replication.[40][41] After translesion synthesis, extension is required. This extension can be carried out by a replicative polymerase if the TLS is error-free, as in the case of Pol η, yet if TLS results in a mismatch, a specialized polymerase is needed to extend it; Pol ζ. Pol ζ is unique in that it can extend terminal mismatches, whereas more processive polymerases cannot. So when a lesion is encountered, the replication fork will stall, PCNA will switch from a processive polymerase to a TLS polymerase such as Pol ι to fix the lesion, then PCNA may switch to Pol ζ to extend the mismatch, and last PCNA will switch to the processive polymerase to continue replication.
Global response to DNA damage[edit]
Cells exposed to ionizing radiation, ultraviolet light or chemicals are prone to acquire multiple sites of bulky DNA lesions and double-strand breaks. Moreover, DNA damaging agents can damage other biomolecules such as proteins, carbohydrates, lipids, and RNA. The accumulation of damage, to be specific, double-strand breaks or adducts stalling the replication forks, are among known stimulation signals for a global response to DNA damage.[42] The global response to damage is an act directed toward the cells’ own preservation and triggers multiple pathways of macromolecular repair, lesion bypass, tolerance, or apoptosis. The common features of global response are induction of multiple genes, cell cycle arrest, and inhibition of cell division.
Initial steps[edit]
The packaging of eukaryotic DNA into chromatin presents a barrier to all DNA-based processes that require recruitment of enzymes to their sites of action. To allow DNA repair, the chromatin must be remodeled. In eukaryotes, ATP dependent chromatin remodeling complexes and histone-modifying enzymes are two predominant factors employed to accomplish this remodeling process.[43]
Chromatin relaxation occurs rapidly at the site of a DNA damage.[44][45] In one of the earliest steps, the stress-activated protein kinase, c-Jun N-terminal kinase (JNK), phosphorylates SIRT6 on serine 10 in response to double-strand breaks or other DNA damage.[46] This post-translational modification facilitates the mobilization of SIRT6 to DNA damage sites, and is required for efficient recruitment of poly (ADP-ribose) polymerase 1 (PARP1) to DNA break sites and for efficient repair of DSBs.[46] PARP1 protein starts to appear at DNA damage sites in less than a second, with half maximum accumulation within 1.6 seconds after the damage occurs.[47] PARP1 synthesizes polymeric adenosine diphosphate ribose (poly (ADP-ribose) or PAR) chains on itself. Next the chromatin remodeler ALC1 quickly attaches to the product of PARP1 action, a poly-ADP ribose chain, and ALC1 completes arrival at the DNA damage within 10 seconds of the occurrence of the damage.[45] About half of the maximum chromatin relaxation, presumably due to action of ALC1, occurs by 10 seconds.[45] This then allows recruitment of the DNA repair enzyme MRE11, to initiate DNA repair, within 13 seconds.[47]
γH2AX, the phosphorylated form of H2AX is also involved in the early steps leading to chromatin decondensation after DNA double-strand breaks. The histone variant H2AX constitutes about 10% of the H2A histones in human chromatin.[48] γH2AX (H2AX phosphorylated on serine 139) can be detected as soon as 20 seconds after irradiation of cells (with DNA double-strand break formation), and half maximum accumulation of γH2AX occurs in one minute.[48] The extent of chromatin with phosphorylated γH2AX is about two million base pairs at the site of a DNA double-strand break.[48] γH2AX does not, itself, cause chromatin decondensation, but within 30 seconds of irradiation, RNF8 protein can be detected in association with γH2AX.[49] RNF8 mediates extensive chromatin decondensation, through its subsequent interaction with CHD4,[50] a component of the nucleosome remodeling and deacetylase complex NuRD.
DDB2 occurs in a heterodimeric complex with DDB1. This complex further complexes with the ubiquitin ligase protein CUL4A[51] and with PARP1.[52] This larger complex rapidly associates with UV-induced damage within chromatin, with half-maximum association completed in 40 seconds.[51] The PARP1 protein, attached to both DDB1 and DDB2, then PARylates (creates a poly-ADP ribose chain) on DDB2 that attracts the DNA remodeling protein ALC1.[52] Action of ALC1 relaxes the chromatin at the site of UV damage to DNA. This relaxation allows other proteins in the nucleotide excision repair pathway to enter the chromatin and repair UV-induced cyclobutane pyrimidine dimer damages.
After rapid chromatin remodeling, cell cycle checkpoints are activated to allow DNA repair to occur before the cell cycle progresses. First, two kinases, ATM and ATR are activated within 5 or 6 minutes after DNA is damaged. This is followed by phosphorylation of the cell cycle checkpoint protein Chk1, initiating its function, about 10 minutes after DNA is damaged.[53]
DNA damage checkpoints[edit]
After DNA damage, cell cycle checkpoints are activated. Checkpoint activation pauses the cell cycle and gives the cell time to repair the damage before continuing to divide. DNA damage checkpoints occur at the G1/S and G2/M boundaries. An intra-S checkpoint also exists. Checkpoint activation is controlled by two master kinases, ATM and ATR. ATM responds to DNA double-strand breaks and disruptions in chromatin structure,[54] whereas ATR primarily responds to stalled replication forks. These kinases phosphorylate downstream targets in a signal transduction cascade, eventually leading to cell cycle arrest. A class of checkpoint mediator proteins including BRCA1, MDC1, and 53BP1 has also been identified.[55] These proteins seem to be required for transmitting the checkpoint activation signal to downstream proteins.
DNA damage checkpoint is a signal transduction pathway that blocks cell cycle progression in G1, G2 and metaphase and slows down the rate of S phase progression when DNA is damaged. It leads to a pause in cell cycle allowing the cell time to repair the damage before continuing to divide.
Checkpoint Proteins can be separated into four groups: phosphatidylinositol 3-kinase (PI3K)-like protein kinase, proliferating cell nuclear antigen (PCNA)-like group, two serine/threonine(S/T) kinases and their adaptors. Central to all DNA damage induced checkpoints responses is a pair of large protein kinases belonging to the first group of PI3K-like protein kinases-the ATM (Ataxia telangiectasia mutated) and ATR (Ataxia- and Rad-related) kinases, whose sequence and functions have been well conserved in evolution. All DNA damage response requires either ATM or ATR because they have the ability to bind to the chromosomes at the site of DNA damage, together with accessory proteins that are platforms on which DNA damage response components and DNA repair complexes can be assembled.
An important downstream target of ATM and ATR is p53, as it is required for inducing apoptosis following DNA damage.[56] The cyclin-dependent kinase inhibitor p21 is induced by both p53-dependent and p53-independent mechanisms and can arrest the cell cycle at the G1/S and G2/M checkpoints by deactivating cyclin/cyclin-dependent kinase complexes.[57]
The prokaryotic SOS response[edit]
The SOS response is the changes in gene expression in Escherichia coli and other bacteria in response to extensive DNA damage. The prokaryotic SOS system is regulated by two key proteins: LexA and RecA. The LexA homodimer is a transcriptional repressor that binds to operator sequences commonly referred to as SOS boxes. In Escherichia coli it is known that LexA regulates transcription of approximately 48 genes including the lexA and recA genes.[58] The SOS response is known to be widespread in the Bacteria domain, but it is mostly absent in some bacterial phyla, like the Spirochetes.[59]
The most common cellular signals activating the SOS response are regions of single-stranded DNA (ssDNA), arising from stalled replication forks or double-strand breaks, which are processed by DNA helicase to separate the two DNA strands.[42] In the initiation step, RecA protein binds to ssDNA in an ATP hydrolysis driven reaction creating RecA–ssDNA filaments. RecA–ssDNA filaments activate LexA autoprotease activity, which ultimately leads to cleavage of LexA dimer and subsequent LexA degradation. The loss of LexA repressor induces transcription of the SOS genes and allows for further signal induction, inhibition of cell division and an increase in levels of proteins responsible for damage processing.
In Escherichia coli, SOS boxes are 20-nucleotide long sequences near promoters with palindromic structure and a high degree of sequence conservation. In other classes and phyla, the sequence of SOS boxes varies considerably, with different length and composition, but it is always highly conserved and one of the strongest short signals in the genome.[59] The high information content of SOS boxes permits differential binding of LexA to different promoters and allows for timing of the SOS response. The lesion repair genes are induced at the beginning of SOS response. The error-prone translesion polymerases, for example, UmuCD’2 (also called DNA polymerase V), are induced later on as a last resort.[60] Once the DNA damage is repaired or bypassed using polymerases or through recombination, the amount of single-stranded DNA in cells is decreased, lowering the amounts of RecA filaments decreases cleavage activity of LexA homodimer, which then binds to the SOS boxes near promoters and restores normal gene expression.
Eukaryotic transcriptional responses to DNA damage[edit]
Eukaryotic cells exposed to DNA damaging agents also activate important defensive pathways by inducing multiple proteins involved in DNA repair, cell cycle checkpoint control, protein trafficking and degradation. Such genome wide transcriptional response is very complex and tightly regulated, thus allowing coordinated global response to damage. Exposure of yeast Saccharomyces cerevisiae to DNA damaging agents results in overlapping but distinct transcriptional profiles. Similarities to environmental shock response indicates that a general global stress response pathway exist at the level of transcriptional activation. In contrast, different human cell types respond to damage differently indicating an absence of a common global response. The probable explanation for this difference between yeast and human cells may be in the heterogeneity of mammalian cells. In an animal different types of cells are distributed among different organs that have evolved different sensitivities to DNA damage.[61]
In general global response to DNA damage involves expression of multiple genes responsible for postreplication repair, homologous recombination, nucleotide excision repair, DNA damage checkpoint, global transcriptional activation, genes controlling mRNA decay, and many others. A large amount of damage to a cell leaves it with an important decision: undergo apoptosis and die, or survive at the cost of living with a modified genome. An increase in tolerance to damage can lead to an increased rate of survival that will allow a greater accumulation of mutations. Yeast Rev1 and human polymerase η are members of Y family translesion DNA polymerases present during global response to DNA damage and are responsible for enhanced mutagenesis during a global response to DNA damage in eukaryotes.[42]
Aging[edit]
Pathological effects of poor DNA repair[edit]

DNA repair rate is an important determinant of cell pathology.
Experimental animals with genetic deficiencies in DNA repair often show decreased life span and increased cancer incidence.[15] For example, mice deficient in the dominant NHEJ pathway and in telomere maintenance mechanisms get lymphoma and infections more often, and, as a consequence, have shorter lifespans than wild-type mice.[62] In similar manner, mice deficient in a key repair and transcription protein that unwinds DNA helices have premature onset of aging-related diseases and consequent shortening of lifespan.[63] However, not every DNA repair deficiency creates exactly the predicted effects; mice deficient in the NER pathway exhibited shortened life span without correspondingly higher rates of mutation.[64]
If the rate of DNA damage exceeds the capacity of the cell to repair it, the accumulation of errors can overwhelm the cell and result in early senescence, apoptosis, or cancer. Inherited diseases associated with faulty DNA repair functioning result in premature aging,[15] increased sensitivity to carcinogens and correspondingly increased cancer risk (see below). On the other hand, organisms with enhanced DNA repair systems, such as Deinococcus radiodurans, the most radiation-resistant known organism, exhibit remarkable resistance to the double-strand break-inducing effects of radioactivity, likely due to enhanced efficiency of DNA repair and especially NHEJ.[65]
Longevity and caloric restriction[edit]

Most life span influencing genes affect the rate of DNA damage.
A number of individual genes have been identified as influencing variations in life span within a population of organisms. The effects of these genes is strongly dependent on the environment, in particular, on the organism’s diet. Caloric restriction reproducibly results in extended lifespan in a variety of organisms, likely via nutrient sensing pathways and decreased metabolic rate. The molecular mechanisms by which such restriction results in lengthened lifespan are as yet unclear (see[66] for some discussion); however, the behavior of many genes known to be involved in DNA repair is altered under conditions of caloric restriction. Several agents reported to have anti-aging properties have been shown to attenuate constitutive level of mTOR signaling, an evidence of reduction of metabolic activity, and concurrently to reduce constitutive level of DNA damage induced by endogenously generated reactive oxygen species.[67]
For example, increasing the gene dosage of the gene SIR-2, which regulates DNA packaging in the nematode worm Caenorhabditis elegans, can significantly extend lifespan.[68] The mammalian homolog of SIR-2 is known to induce downstream DNA repair factors involved in NHEJ, an activity that is especially promoted under conditions of caloric restriction.[69] Caloric restriction has been closely linked to the rate of base excision repair in the nuclear DNA of rodents,[70] although similar effects have not been observed in mitochondrial DNA.[71]
The C. elegans gene AGE-1, an upstream effector of DNA repair pathways, confers dramatically extended life span under free-feeding conditions but leads to a decrease in reproductive fitness under conditions of caloric restriction.[72] This observation supports the pleiotropy theory of the biological origins of aging, which suggests that genes conferring a large survival advantage early in life will be selected for even if they carry a corresponding disadvantage late in life.
Medicine and DNA repair modulation[edit]
Hereditary DNA repair disorders[edit]
Defects in the NER mechanism are responsible for several genetic disorders, including:
- Xeroderma pigmentosum: hypersensitivity to sunlight/UV, resulting in increased skin cancer incidence and premature aging
- Cockayne syndrome: hypersensitivity to UV and chemical agents
- Trichothiodystrophy: sensitive skin, brittle hair and nails
Mental retardation often accompanies the latter two disorders, suggesting increased vulnerability of developmental neurons.
Other DNA repair disorders include:
- Werner’s syndrome: premature aging and retarded growth
- Bloom’s syndrome: sunlight hypersensitivity, high incidence of malignancies (especially leukemias).
- Ataxia telangiectasia: sensitivity to ionizing radiation and some chemical agents
All of the above diseases are often called «segmental progerias» («accelerated aging diseases») because those affected appear elderly and experience aging-related diseases at an abnormally young age, while not manifesting all the symptoms of old age.
Other diseases associated with reduced DNA repair function include Fanconi anemia, hereditary breast cancer and hereditary colon cancer.
Cancer[edit]
Because of inherent limitations in the DNA repair mechanisms, if humans lived long enough, they would all eventually develop cancer.[73][74] There are at least 34 Inherited human DNA repair gene mutations that increase cancer risk. Many of these mutations cause DNA repair to be less effective than normal. In particular, Hereditary nonpolyposis colorectal cancer (HNPCC) is strongly associated with specific mutations in the DNA mismatch repair pathway. BRCA1 and BRCA2, two important genes whose mutations confer a hugely increased risk of breast cancer on carriers,[75] are both associated with a large number of DNA repair pathways, especially NHEJ and homologous recombination.
Cancer therapy procedures such as chemotherapy and radiotherapy work by overwhelming the capacity of the cell to repair DNA damage, resulting in cell death. Cells that are most rapidly dividing – most typically cancer cells – are preferentially affected. The side-effect is that other non-cancerous but rapidly dividing cells such as progenitor cells in the gut, skin, and hematopoietic system are also affected. Modern cancer treatments attempt to localize the DNA damage to cells and tissues only associated with cancer, either by physical means (concentrating the therapeutic agent in the region of the tumor) or by biochemical means (exploiting a feature unique to cancer cells in the body). In the context of therapies targeting DNA damage response genes, the latter approach has been termed ‘synthetic lethality’.[76]
Perhaps the most well-known of these ‘synthetic lethality’ drugs is the poly(ADP-ribose) polymerase 1 (PARP1) inhibitor olaparib, which was approved by the Food and Drug Administration in 2015 for the treatment in women of BRCA-defective ovarian cancer. Tumor cells with partial loss of DNA damage response (specifically, homologous recombination repair) are dependent on another mechanism – single-strand break repair – which is a mechanism consisting, in part, of the PARP1 gene product.[77] Olaparib is combined with chemotherapeutics to inhibit single-strand break repair induced by DNA damage caused by the co-administered chemotherapy. Tumor cells relying on this residual DNA repair mechanism are unable to repair the damage and hence are not able to survive and proliferate, whereas normal cells can repair the damage with the functioning homologous recombination mechanism.
Many other drugs for use against other residual DNA repair mechanisms commonly found in cancer are currently under investigation. However, synthetic lethality therapeutic approaches have been questioned due to emerging evidence of acquired resistance, achieved through rewiring of DNA damage response pathways and reversion of previously inhibited defects.[78]
DNA repair defects in cancer[edit]
It has become apparent over the past several years that the DNA damage response acts as a barrier to the malignant transformation of preneoplastic cells.[79] Previous studies have shown an elevated DNA damage response in cell-culture models with oncogene activation[80] and preneoplastic colon adenomas.[81] DNA damage response mechanisms trigger cell-cycle arrest, and attempt to repair DNA lesions or promote cell death/senescence if repair is not possible. Replication stress is observed in preneoplastic cells due to increased proliferation signals from oncogenic mutations. Replication stress is characterized by: increased replication initiation/origin firing; increased transcription and collisions of transcription-replication complexes; nucleotide deficiency; increase in reactive oxygen species (ROS).[82]
Replication stress, along with the selection for inactivating mutations in DNA damage response genes in the evolution of the tumor,[83] leads to downregulation and/or loss of some DNA damage response mechanisms, and hence loss of DNA repair and/or senescence/programmed cell death. In experimental mouse models, loss of DNA damage response-mediated cell senescence was observed after using a short hairpin RNA (shRNA) to inhibit the double-strand break response kinase ataxia telangiectasia (ATM), leading to increased tumor size and invasiveness.[81] Humans born with inherited defects in DNA repair mechanisms (for example, Li-Fraumeni syndrome) have a higher cancer risk.[84]
The prevalence of DNA damage response mutations differs across cancer types; for example, 30% of breast invasive carcinomas have mutations in genes involved in homologous recombination.[79] In cancer, downregulation is observed across all DNA damage response mechanisms (base excision repair (BER), nucleotide excision repair (NER), DNA mismatch repair (MMR), homologous recombination repair (HR), non-homologous end joining (NHEJ) and translesion DNA synthesis (TLS).[85] As well as mutations to DNA damage repair genes, mutations also arise in the genes responsible for arresting the cell cycle to allow sufficient time for DNA repair to occur, and some genes are involved in both DNA damage repair and cell cycle checkpoint control, for example ATM and checkpoint kinase 2 (CHEK2) – a tumor suppressor that is often absent or downregulated in non-small cell lung cancer.[86]
Genes involved in DNA damage response pathways and frequently mutated in cancer (HR = homologous recombination; NHEJ = non-homologous end joining; SSA = single-strand annealing; FA = fanconi anemia pathway; BER = base excision repair; NER = nucleotide excision repair; MMR = mismatch repair)
| HR | NHEJ | SSA | FA | BER | NER | MMR | |
|---|---|---|---|---|---|---|---|
| ATM | |||||||
| ATR | |||||||
| PAXIP | |||||||
| RPA | |||||||
| BRCA1 | |||||||
| BRCA2 | |||||||
| RAD51 | |||||||
| RFC | |||||||
| XRCC1 | |||||||
| PCNA | |||||||
| PARP1 | |||||||
| ERCC1 | |||||||
| MSH3 |
Epigenetic DNA repair defects in cancer[edit]
Classically, cancer has been viewed as a set of diseases that are driven by progressive genetic abnormalities that include mutations in tumour-suppressor genes and oncogenes, and chromosomal aberrations. However, it has become apparent that cancer is also driven by
epigenetic alterations.[87]
Epigenetic alterations refer to functionally relevant modifications to the genome that do not involve a change in the nucleotide sequence. Examples of such modifications are changes in DNA methylation (hypermethylation and hypomethylation) and histone modification,[88] changes in chromosomal architecture (caused by inappropriate expression of proteins such as HMGA2 or HMGA1)[89] and changes caused by microRNAs. Each of these epigenetic alterations serves to regulate gene expression without altering the underlying DNA sequence. These changes usually remain through cell divisions, last for multiple cell generations, and can be considered to be epimutations (equivalent to mutations).
While large numbers of epigenetic alterations are found in cancers, the epigenetic alterations in DNA repair genes, causing reduced expression of DNA repair proteins, appear to be particularly important. Such alterations are thought to occur early in progression to cancer and to be a likely cause of the genetic instability characteristic of cancers.[90][91][92]
Reduced expression of DNA repair genes causes deficient DNA repair. When DNA repair is deficient DNA damages remain in cells at a higher than usual level and these excess damages cause increased frequencies of mutation or epimutation. Mutation rates increase substantially in cells defective in DNA mismatch repair[93][94] or in homologous recombinational repair (HRR).[95] Chromosomal rearrangements and aneuploidy also increase in HRR defective cells.[96]
Higher levels of DNA damage not only cause increased mutation, but also cause increased epimutation. During repair of DNA double strand breaks, or repair of other DNA damages, incompletely cleared sites of repair can cause epigenetic gene silencing.[97][98]
Deficient expression of DNA repair proteins due to an inherited mutation can cause increased risk of cancer. Individuals with an inherited impairment in any of 34 DNA repair genes (see article DNA repair-deficiency disorder) have an increased risk of cancer, with some defects causing up to a 100% lifetime chance of cancer (e.g. p53 mutations).[99] However, such germline mutations (which cause highly penetrant cancer syndromes) are the cause of only about 1 percent of cancers.[100]
Frequencies of epimutations in DNA repair genes[edit]

A chart of common DNA damaging agents, examples of lesions they cause in DNA, and pathways used to repair these lesions. Also shown are many of the genes in these pathways, an indication of which genes are epigenetically regulated to have reduced (or increased) expression in various cancers. It also shows genes in the error-prone microhomology-mediated end joining pathway with increased expression in various cancers.
Deficiencies in DNA repair enzymes are occasionally caused by a newly arising somatic mutation in a DNA repair gene, but are much more frequently caused by epigenetic alterations that reduce or silence expression of DNA repair genes. For example, when 113 colorectal cancers were examined in sequence, only four had a missense mutation in the DNA repair gene MGMT, while the majority had reduced MGMT expression due to methylation of the MGMT promoter region (an epigenetic alteration).[101] Five different studies found that between 40% and 90% of colorectal cancers have reduced MGMT expression due to methylation of the MGMT promoter region.[102][103][104][105][106]
Similarly, out of 119 cases of mismatch repair-deficient colorectal cancers that lacked DNA repair gene PMS2 expression, PMS2 was deficient in 6 due to mutations in the PMS2 gene, while in 103 cases PMS2 expression was deficient because its pairing partner MLH1 was repressed due to promoter methylation (PMS2 protein is unstable in the absence of MLH1).[107] In the other 10 cases, loss of PMS2 expression was likely due to epigenetic overexpression of the microRNA, miR-155, which down-regulates MLH1.[108]
In a further example, epigenetic defects were found in various cancers (e.g. breast, ovarian, colorectal and head and neck). Two or three deficiencies in the expression of ERCC1, XPF or PMS2 occur simultaneously in the majority of 49 colon cancers evaluated by Facista et al.[109]
The chart in this section shows some frequent DNA damaging agents, examples of DNA lesions they cause, and the pathways that deal with these DNA damages. At least 169 enzymes are either directly employed in DNA repair or influence DNA repair processes.[110] Of these, 83 are directly employed in repairing the 5 types of DNA damages illustrated in the chart.
Some of the more well studied genes central to these repair processes are shown in the chart. The gene designations shown in red, gray or cyan indicate genes frequently epigenetically altered in various types of cancers. Wikipedia articles on each of the genes highlighted by red, gray or cyan describe the epigenetic alteration(s) and the cancer(s) in which these epimutations are found. Review articles,[111] and broad experimental survey articles[112][113] also document most of these epigenetic DNA repair deficiencies in cancers.
Red-highlighted genes are frequently reduced or silenced by epigenetic mechanisms in various cancers. When these genes have low or absent expression, DNA damages can accumulate. Replication errors past these damages (see translesion synthesis) can lead to increased mutations and, ultimately, cancer. Epigenetic repression of DNA repair genes in accurate DNA repair pathways appear to be central to carcinogenesis.
The two gray-highlighted genes RAD51 and BRCA2, are required for homologous recombinational repair. They are sometimes epigenetically over-expressed and sometimes under-expressed in certain cancers. As indicated in the Wikipedia articles on RAD51 and BRCA2, such cancers ordinarily have epigenetic deficiencies in other DNA repair genes. These repair deficiencies would likely cause increased unrepaired DNA damages. The over-expression of RAD51 and BRCA2 seen in these cancers may reflect selective pressures for compensatory RAD51 or BRCA2 over-expression and increased homologous recombinational repair to at least partially deal with such excess DNA damages. In those cases where RAD51 or BRCA2 are under-expressed, this would itself lead to increased unrepaired DNA damages. Replication errors past these damages (see translesion synthesis) could cause increased mutations and cancer, so that under-expression of RAD51 or BRCA2 would be carcinogenic in itself.
Cyan-highlighted genes are in the microhomology-mediated end joining (MMEJ) pathway and are up-regulated in cancer. MMEJ is an additional error-prone inaccurate repair pathway for double-strand breaks. In MMEJ repair of a double-strand break, an homology of 5–25 complementary base pairs between both paired strands is sufficient to align the strands, but mismatched ends (flaps) are usually present. MMEJ removes the extra nucleotides (flaps) where strands are joined, and then ligates the strands to create an intact DNA double helix. MMEJ almost always involves at least a small deletion, so that it is a mutagenic pathway.[24] FEN1, the flap endonuclease in MMEJ, is epigenetically increased by promoter hypomethylation and is over-expressed in the majority of cancers of the breast,[114] prostate,[115] stomach,[116][117] neuroblastomas,[118] pancreas,[119] and lung.[120] PARP1 is also over-expressed when its promoter region ETS site is epigenetically hypomethylated, and this contributes to progression to endometrial cancer[121] and BRCA-mutated serous ovarian cancer.[122] Other genes in the MMEJ pathway are also over-expressed in a number of cancers (see MMEJ for summary), and are also shown in cyan.
Genome-wide distribution of DNA repair in human somatic cells[edit]
Differential activity of DNA repair pathways across various regions of the human genome causes mutations to be very unevenly distributed within tumor genomes.[123][124] In particular, the gene-rich, early-replicating regions of the human genome exhibit lower mutation frequencies than the gene-poor, late-replicating heterochromatin. One mechanism underlying this involves the histone modification H3K36me3, which can recruit mismatch repair proteins,[125] thereby lowering mutation rates in H3K36me3-marked regions.[126] Another important mechanism concerns nucleotide excision repair, which can be recruited by the transcription machinery, lowering somatic mutation rates in active genes[124] and other open chromatin regions.[127]
Epigenetic alterations due to DNA repair[edit]
Damage to DNA is very common and is constantly being repaired. Epigenetic alterations can accompany DNA repair of oxidative damage or double-strand breaks. In human cells, oxidative DNA damage occurs about 10,000 times a day and DNA double-strand breaks occur about 10 to 50 times a cell cycle in somatic replicating cells (see DNA damage (naturally occurring)). The selective advantage of DNA repair is to allow the cell to survive in the face of DNA damage. The selective advantage of epigenetic alterations that occur with DNA repair is not clear.
Repair of oxidative DNA damage can alter epigenetic markers[edit]
In the steady state (with endogenous damages occurring and being repaired), there are about 2,400 oxidatively damaged guanines that form 8-oxo-2′-deoxyguanosine (8-OHdG) in the average mamalian cell DNA.[128] 8-OHdG constitutes about 5% of the oxidative damages commonly present in DNA.[129] The oxidized guanines do not occur randomly among all guanines in DNA. There is a sequence preference for the guanine at a methylated CpG site (a cytosine followed by guanine along its 5′ → 3′ direction and where the cytosine is methylated (5-mCpG)).[130] A 5-mCpG site has the lowest ionization potential for guanine oxidation.

Initiation of DNA demethylation at a CpG site. In adult somatic cells DNA methylation typically occurs in the context of CpG dinucleotides (CpG sites), forming 5-methylcytosine-pG, or 5mCpG. Reactive oxygen species (ROS) may attack guanine at the dinucleotide site, forming 8-hydroxy-2′-deoxyguanosine (8-OHdG), and resulting in a 5mCp-8-OHdG dinucleotide site. The base excision repair enzyme OGG1 targets 8-OHdG and binds to the lesion without immediate excision. OGG1, present at a 5mCp-8-OHdG site recruits TET1 and TET1 oxidizes the 5mC adjacent to the 8-OHdG. This initiates demethylation of 5mC.[131]
Oxidized guanine has mispairing potential and is mutagenic.[132] Oxoguanine glycosylase (OGG1) is the primary enzyme responsible for the excision of the oxidized guanine during DNA repair. OGG1 finds and binds to an 8-OHdG within a few seconds.[133] However, OGG1 does not immediately excise 8-OHdG. In HeLa cells half maximum removal of 8-OHdG occurs in 30 minutes,[134] and in irradiated mice, the 8-OHdGs induced in the mouse liver are removed with a half-life of 11 minutes.[129]
When OGG1 is present at an oxidized guanine within a methylated CpG site it recruits TET1 to the 8-OHdG lesion (see Figure). This allows TET1 to demethylate an adjacent methylated cytosine. Demethylation of cytosine is an epigenetic alteration.
As an example, when human mammary epithelial cells were treated with H2O2 for six hours, 8-OHdG increased about 3.5-fold in DNA and this caused about 80% demethylation of the 5-methylcytosines in the genome.[131] Demethylation of CpGs in a gene promoter by TET enzyme activity increases transcription of the gene into messenger RNA.[135] In cells treated with H2O2, one particular gene was examined, BACE1.[131] The methylation level of the BACE1 CpG island was reduced (an epigenetic alteration) and this allowed about 6.5 fold increase of expression of BACE1 messenger RNA.
While six-hour incubation with H2O2 causes considerable demethylation of 5-mCpG sites, shorter times of H2O2 incubation appear to promote other epigenetic alterations. Treatment of cells with H2O2 for 30 minutes causes the mismatch repair protein heterodimer MSH2-MSH6 to recruit DNA methyltransferase 1 (DNMT1) to sites of some kinds of oxidative DNA damage.[136] This could cause increased methylation of cytosines (epigenetic alterations) at these locations.
Jiang et al.[137] treated HEK 293 cells with agents causing oxidative DNA damage, (potassium bromate (KBrO3) or potassium chromate (K2CrO4)). Base excision repair (BER) of oxidative damage occurred with the DNA repair enzyme polymerase beta localizing to oxidized guanines. Polymerase beta is the main human polymerase in short-patch BER of oxidative DNA damage. Jiang et al.[137] also found that polymerase beta recruited the DNA methyltransferase protein DNMT3b to BER repair sites. They then evaluated the methylation pattern at the single nucleotide level in a small region of DNA including the promoter region and the early transcription region of the BRCA1 gene. Oxidative DNA damage from bromate modulated the DNA methylation pattern (caused epigenetic alterations) at CpG sites within the region of DNA studied. In untreated cells, CpGs located at −189, −134, −29, −19, +16, and +19 of the BRCA1 gene had methylated cytosines (where numbering is from the messenger RNA transcription start site, and negative numbers indicate nucleotides in the upstream promoter region). Bromate treatment-induced oxidation resulted in the loss of cytosine methylation at −189, −134, +16 and +19 while also leading to the formation of new methylation at the CpGs located at −80, −55, −21 and +8 after DNA repair was allowed.
Homologous recombinational repair alters epigenetic markers[edit]
At least four articles report the recruitment of DNA methyltransferase 1 (DNMT1) to sites of DNA double-strand breaks.[138][139][140][141] During homologous recombinational repair (HR) of the double-strand break, the involvement of DNMT1 causes the two repaired strands of DNA to have different levels of methylated cytosines. One strand becomes frequently methylated at about 21 CpG sites downstream of the repaired double-strand break. The other DNA strand loses methylation at about six CpG sites that were previously methylated downstream of the double-strand break, as well as losing methylation at about five CpG sites that were previously methylated upstream of the double-strand break. When the chromosome is replicated, this gives rise to one daughter chromosome that is heavily methylated downstream of the previous break site and one that is unmethylated in the region both upstream and downstream of the previous break site. With respect to the gene that was broken by the double-strand break, half of the progeny cells express that gene at a high level and in the other half of the progeny cells expression of that gene is repressed. When clones of these cells were maintained for three years, the new methylation patterns were maintained over that time period.[142]
In mice with a CRISPR-mediated homology-directed recombination insertion in their genome there were a large number of increased methylations of CpG sites within the double-strand break-associated insertion.[143]
Non-homologous end joining can cause some epigenetic marker alterations[edit]
Non-homologous end joining (NHEJ) repair of a double-strand break can cause a small number of demethylations of pre-existing cytosine DNA methylations downstream of the repaired double-strand break.[139] Further work by Allen et al.[144] showed that NHEJ of a DNA double-strand break in a cell could give rise to some progeny cells having repressed expression of the gene harboring the initial double-strand break and some progeny having high expression of that gene due to epigenetic alterations associated with NHEJ repair. The frequency of epigenetic alterations causing repression of a gene after an NHEJ repair of a DNA double-strand break in that gene may be about 0.9%.[140]
Evolution[edit]
The basic processes of DNA repair are highly conserved among both prokaryotes and eukaryotes and even among bacteriophages (viruses which infect bacteria); however, more complex organisms with more complex genomes have correspondingly more complex repair mechanisms.[145] The ability of a large number of protein structural motifs to catalyze relevant chemical reactions has played a significant role in the elaboration of repair mechanisms during evolution. For an extremely detailed review of hypotheses relating to the evolution of DNA repair, see.[146]
The fossil record indicates that single-cell life began to proliferate on the planet at some point during the Precambrian period, although exactly when recognizably modern life first emerged is unclear. Nucleic acids became the sole and universal means of encoding genetic information, requiring DNA repair mechanisms that in their basic form have been inherited by all extant life forms from their common ancestor. The emergence of Earth’s oxygen-rich atmosphere (known as the «oxygen catastrophe») due to photosynthetic organisms, as well as the presence of potentially damaging free radicals in the cell due to oxidative phosphorylation, necessitated the evolution of DNA repair mechanisms that act specifically to counter the types of damage induced by oxidative stress.
Rate of evolutionary change[edit]
On some occasions, DNA damage is not repaired or is repaired by an error-prone mechanism that results in a change from the original sequence. When this occurs, mutations may propagate into the genomes of the cell’s progeny. Should such an event occur in a germ line cell that will eventually produce a gamete, the mutation has the potential to be passed on to the organism’s offspring. The rate of evolution in a particular species (or, in a particular gene) is a function of the rate of mutation. As a consequence, the rate and accuracy of DNA repair mechanisms have an influence over the process of evolutionary change.[147] DNA damage protection and repair does not influence the rate of adaptation by gene regulation and by recombination and selection of alleles. On the other hand, DNA damage repair and protection does influence the rate of accumulation of irreparable, advantageous, code expanding, inheritable mutations, and slows down the evolutionary mechanism for expansion of the genome of organisms with new functionalities. The tension between evolvability and mutation repair and protection needs further investigation.
Technology[edit]
A technology named clustered regularly interspaced short palindromic repeat (shortened to CRISPR-Cas9) was discovered in 2012. The new technology allows anyone with molecular biology training to alter the genes of any species with precision, by inducing DNA damage at a specific point and then altering DNA repair mechanisms to insert new genes.[148] It is cheaper, more efficient, and more precise than other technologies. With the help of CRISPR–Cas9, parts of a genome can be edited by scientists by removing, adding, or altering parts in a DNA sequence.
See also[edit]
- Accelerated aging disease
- Aging DNA
- Cell cycle
- DNA damage (naturally occurring)
- DNA damage theory of aging
- DNA replication
- Direct DNA damage
- Gene therapy
- Human mitochondrial genetics
- Indirect DNA damage
- Life extension
- Progeria
- REPAIRtoire
- Senescence
- SiDNA
- The scientific journal DNA Repair under Mutation Research
References[edit]
- ^ «Nature Reviews Series: DNA damage». Nature Reviews Molecular Cell Biology. 5 July 2017. Retrieved 7 November 2018.
- ^ a b Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J (2004). Molecular Biology of the Cell (5th ed.). New York: WH Freeman. p. 963.
- ^ a b Acharya PV (1971). «The isolation and partial characterization of age-correlated oligo-deoxyribo-ribonucleotides with covalently linked aspartyl-glutamyl polypeptides». Johns Hopkins Medical Journal. Supplement (1): 254–60. PMID 5055816.
- ^ a b Bjorksten J, Acharya PV, Ashman S, Wetlaufer DB (July 1971). «Gerogenic fractions in the tritiated rat». Journal of the American Geriatrics Society. 19 (7): 561–74. doi:10.1111/j.1532-5415.1971.tb02577.x. PMID 5106728. S2CID 33154242.
- ^ Browner WS, Kahn AJ, Ziv E, Reiner AP, Oshima J, Cawthon RM, et al. (December 2004). «The genetics of human longevity». The American Journal of Medicine. 117 (11): 851–60. CiteSeerX 10.1.1.556.6874. doi:10.1016/j.amjmed.2004.06.033. PMID 15589490.
- ^ Broad WJ (7 October 2015). «Nobel Prize in Chemistry Awarded to Tomas Lindahl, Paul Modrich and Aziz Sancar for DNA Studies». The New York Times. Retrieved 7 October 2015.
- ^ Staff (7 October 2015). «The Nobel Prize in Chemistry 2015 – DNA repair – providing chemical stability for life» (PDF). Nobel Prize. Retrieved 7 October 2015.
- ^ Roulston A, Marcellus RC, Branton PE (1999). «Viruses and apoptosis». Annual Review of Microbiology. 53: 577–628. doi:10.1146/annurev.micro.53.1.577. PMID 10547702.
- ^ Madigan MT, Martino JM (2006). Brock Biology of Microorganisms (11th ed.). Pearson. p. 136. ISBN 978-0-13-196893-6.
- ^ Ohta T, Tokishita SI, Mochizuki K, Kawase J, Sakahira M, Yamagata H (2006). «UV Sensitivity and Mutagenesis of the Extremely Thermophilic Eubacterium Thermus thermophilus HB27». Genes and Environment. 28 (2): 56–61. doi:10.3123/jemsge.28.56.
- ^ Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z (September 2006). «Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants». Cell Cycle. 5 (17): 1940–45. doi:10.4161/cc.5.17.3191. PMC 3488278. PMID 16940754.
- ^ Braig M, Schmitt CA (March 2006). «Oncogene-induced senescence: putting the brakes on tumor development». Cancer Research. 66 (6): 2881–84. doi:10.1158/0008-5472.CAN-05-4006. PMID 16540631.
- ^ Lynch MD (February 2006). «How does cellular senescence prevent cancer?». DNA and Cell Biology. 25 (2): 69–78. doi:10.1089/dna.2006.25.69. PMID 16460230.
- ^ Campisi J, d’Adda di Fagagna F (September 2007). «Cellular senescence: when bad things happen to good cells». Nature Reviews. Molecular Cell Biology. 8 (9): 729–40. doi:10.1038/nrm2233. PMID 17667954. S2CID 15664931.
- ^ a b c Best BP (June 2009). «Nuclear DNA damage as a direct cause of aging» (PDF). Rejuvenation Research. 12 (3): 199–208. CiteSeerX 10.1.1.318.738. doi:10.1089/rej.2009.0847. PMID 19594328. Archived from the original (PDF) on 15 November 2017. Retrieved 29 September 2009.
- ^ Sancar A (June 2003). «Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors». Chemical Reviews. 103 (6): 2203–37. doi:10.1021/cr0204348. PMID 12797829.
- ^ Lucas-Lledó JI, Lynch M (May 2009). «Evolution of mutation rates: phylogenomic analysis of the photolyase/cryptochrome family». Molecular Biology and Evolution. 26 (5): 1143–53. doi:10.1093/molbev/msp029. PMC 2668831. PMID 19228922.
- ^ a b c Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R (2004). Molecular Biology of the Gene (5th ed.). Pearson Benjamin Cummings; CSHL Press. Ch. 9, 10. OCLC 936762772.
- ^ Volkert MR (1988). «Adaptive response of Escherichia coli to alkylation damage». Environmental and Molecular Mutagenesis. 11 (2): 241–55. doi:10.1002/em.2850110210. PMID 3278898. S2CID 24722637.
- ^ a b c Willey J, Sherwood L, Woolverton C (2014). Prescott’s Microbiology. New York: McGraw Hill. p. 381. ISBN 978-0-07-3402-40-6.
- ^ Russell P (2018). i Genetics. Chennai: Pearson. p. 186. ISBN 978-93-325-7162-4.
- ^ a b c d Reardon JT, Sancar A (2006). «Purification and characterization of Escherichia coli and human nucleotide excision repair enzyme systems». Methods in Enzymology. 408: 189–213. doi:10.1016/S0076-6879(06)08012-8. ISBN 9780121828134. PMID 16793370.
- ^ Berg M, Tymoczko J, Stryer L (2012). Biochemistry 7th edition. New York: W.H. Freeman and Company. p. 840. ISBN 9781429229364.
- ^ a b Liang L, Deng L, Chen Y, Li GC, Shao C, Tischfield JA (September 2005). «Modulation of DNA end joining by nuclear proteins». The Journal of Biological Chemistry. 280 (36): 31442–49. doi:10.1074/jbc.M503776200. PMID 16012167.
- ^ Wilson TE, Grawunder U, Lieber MR (July 1997). «Yeast DNA ligase IV mediates non-homologous DNA end joining». Nature. 388 (6641): 495–98. Bibcode:1997Natur.388..495W. doi:10.1038/41365. PMID 9242411. S2CID 4422938.
- ^ Moore JK, Haber JE (May 1996). «Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae». Molecular and Cellular Biology. 16 (5): 2164–73. doi:10.1128/mcb.16.5.2164. PMC 231204. PMID 8628283.
- ^ Boulton SJ, Jackson SP (September 1996). «Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways». The EMBO Journal. 15 (18): 5093–103. doi:10.1002/j.1460-2075.1996.tb00890.x. PMC 452249. PMID 8890183.
- ^ Wilson TE, Lieber MR (August 1999). «Efficient processing of DNA ends during yeast nonhomologous end joining. Evidence for a DNA polymerase beta (Pol4)-dependent pathway». The Journal of Biological Chemistry. 274 (33): 23599–609. doi:10.1074/jbc.274.33.23599. PMID 10438542.
- ^ Budman J, Chu G (February 2005). «Processing of DNA for nonhomologous end-joining by cell-free extract». The EMBO Journal. 24 (4): 849–60. doi:10.1038/sj.emboj.7600563. PMC 549622. PMID 15692565.
- ^ Wang H, Perrault AR, Takeda Y, Qin W, Wang H, Iliakis G (September 2003). «Biochemical evidence for Ku-independent backup pathways of NHEJ». Nucleic Acids Research. 31 (18): 5377–88. doi:10.1093/nar/gkg728. PMC 203313. PMID 12954774.
- ^ Jung D, Alt FW (January 2004). «Unraveling V(D)J recombination; insights into gene regulation». Cell. 116 (2): 299–311. doi:10.1016/S0092-8674(04)00039-X. PMID 14744439. S2CID 16890458.
- ^ a b Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, et al. (May 2013). «Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells». Proceedings of the National Academy of Sciences of the United States of America. 110 (19): 7720–25. Bibcode:2013PNAS..110.7720T. doi:10.1073/pnas.1213431110. PMC 3651503. PMID 23610439.
- ^ Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC (March 2015). «Homology and enzymatic requirements of microhomology-dependent alternative end joining». Cell Death & Disease. 6 (3): e1697. doi:10.1038/cddis.2015.58. PMC 4385936. PMID 25789972.
- ^ Decottignies A (2013). «Alternative end-joining mechanisms: a historical perspective». Frontiers in Genetics. 4: 48. doi:10.3389/fgene.2013.00048. PMC 3613618. PMID 23565119.
- ^ Zahradka K, Slade D, Bailone A, Sommer S, Averbeck D, Petranovic M, et al. (October 2006). «Reassembly of shattered chromosomes in Deinococcus radiodurans». Nature. 443 (7111): 569–73. Bibcode:2006Natur.443..569Z. doi:10.1038/nature05160. PMID 17006450. S2CID 4412830.
- ^ Stenerlöw B, Karlsson KH, Cooper B, Rydberg B. «Measurement of prompt DNA double-strand breaks in mammalian cells without including heat-labile sites: results for cells deficient in nonhomologous end joining». Radiat Res. 2003 Apr;159(4):502–10. doi:10.1667/0033-7587(2003)159[0502:mopdds2.0.co;2] PMID 12643795.
- ^ Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC (March 2009). «Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance». Microbiology and Molecular Biology Reviews. 73 (1): 134–54. doi:10.1128/MMBR.00034-08. PMC 2650891. PMID 19258535.
- ^ Colis LC, Raychaudhury P, Basu AK (August 2008). «Mutational specificity of gamma-radiation-induced guanine-thymine and thymine-guanine intrastrand cross-links in mammalian cells and translesion synthesis past the guanine-thymine lesion by human DNA polymerase eta». Biochemistry. 47 (31): 8070–79. doi:10.1021/bi800529f. PMC 2646719. PMID 18616294.
- ^ Raychaudhury P, Basu AK (March 2011). «Genetic requirement for mutagenesis of the G[8,5-Me]T cross-link in Escherichia coli: DNA polymerases IV and V compete for error-prone bypass». Biochemistry. 50 (12): 2330–38. doi:10.1021/bi102064z. PMC 3062377. PMID 21302943.
- ^ «Translesion Synthesis». Research.chem.psu.edu. Archived from the original on 10 March 2012. Retrieved 14 August 2012.
- ^ Wang Z (July 2001). «Translesion synthesis by the UmuC family of DNA polymerases». Mutation Research. 486 (2): 59–70. doi:10.1016/S0921-8777(01)00089-1. PMID 11425512.
- ^ a b c Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. (2006). DNA Repair and Mutagenesis, part 3. ASM Press. 2nd ed.
- ^ Liu B, Yip RK, Zhou Z (November 2012). «Chromatin remodeling, DNA damage repair and aging». Current Genomics. 13 (7): 533–47. doi:10.2174/138920212803251373. PMC 3468886. PMID 23633913.
- ^ Halicka HD, Zhao H, Podhorecka M, Traganos F, Darzynkiewicz Z (July 2009). «Cytometric detection of chromatin relaxation, an early reporter of DNA damage response». Cell Cycle. 8 (14): 2233–37. doi:10.4161/cc.8.14.8984. PMC 3856216. PMID 19502789.
- ^ a b c Sellou H, Lebeaupin T, Chapuis C, Smith R, Hegele A, Singh HR, et al. (December 2016). «The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage». Molecular Biology of the Cell. 27 (24): 3791–99. doi:10.1091/mbc.E16-05-0269. PMC 5170603. PMID 27733626.
- ^ a b Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, et al. (September 2016). «JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks». Cell Reports. 16 (10): 2641–50. doi:10.1016/j.celrep.2016.08.006. PMC 5089070. PMID 27568560.
- ^ a b Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG (January 2008). «PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites». The Journal of Biological Chemistry. 283 (2): 1197–208. doi:10.1074/jbc.M706734200. PMID 18025084.
- ^ a b c Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (March 1998). «DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139». The Journal of Biological Chemistry. 273 (10): 5858–68. doi:10.1074/jbc.273.10.5858. PMID 9488723.
- ^ Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J (November 2007). «RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins». Cell. 131 (5): 887–900. doi:10.1016/j.cell.2007.09.040. PMID 18001824. S2CID 14232192.
- ^ Luijsterburg MS, Acs K, Ackermann L, Wiegant WW, Bekker-Jensen S, Larsen DH, et al. (May 2012). «A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure». The EMBO Journal. 31 (11): 2511–27. doi:10.1038/emboj.2012.104. PMC 3365417. PMID 22531782.
- ^ a b Luijsterburg MS, Goedhart J, Moser J, Kool H, Geverts B, Houtsmuller AB, et al. (August 2007). «Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC». Journal of Cell Science. 120 (Pt 15): 2706–16. doi:10.1242/jcs.008367. PMID 17635991.
- ^ a b Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, et al. (October 2012). «PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1». The Journal of Cell Biology. 199 (2): 235–49. doi:10.1083/jcb.201112132. PMC 3471223. PMID 23045548.
- ^ Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP (January 2006). «ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks». Nature Cell Biology. 8 (1): 37–45. doi:10.1038/ncb1337. PMID 16327781. S2CID 9797133.
- ^ Bakkenist CJ, Kastan MB (January 2003). «DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation». Nature. 421 (6922): 499–506. Bibcode:2003Natur.421..499B. doi:10.1038/nature01368. PMID 12556884. S2CID 4403303.
- ^ Wei Q, Li L, Chen D (2007). DNA Repair, Genetic Instability, and Cancer. World Scientific. ISBN 978-981-270-014-8.[page needed]
- ^ Schonthal AH (2004). Checkpoint Controls and Cancer. Humana Press. ISBN 978-1-58829-500-2.[page needed]
- ^ Gartel AL, Tyner AL (June 2002). «The role of the cyclin-dependent kinase inhibitor p21 in apoptosis». Molecular Cancer Therapeutics. 1 (8): 639–49. PMID 12479224.
- ^ Janion C (2001). «Some aspects of the SOS response system—a critical survey». Acta Biochimica Polonica. 48 (3): 599–610. doi:10.18388/abp.2001_3894. PMID 11833768.
- ^ a b Erill I, Campoy S, Barbé J (November 2007). «Aeons of distress: an evolutionary perspective on the bacterial SOS response». FEMS Microbiology Reviews. 31 (6): 637–56. doi:10.1111/j.1574-6976.2007.00082.x. PMID 17883408.
- ^ Schlacher K, Pham P, Cox MM, Goodman MF (February 2006). «Roles of DNA polymerase V and RecA protein in SOS damage-induced mutation». Chemical Reviews. 106 (2): 406–19. doi:10.1021/cr0404951. PMID 16464012.
- ^ Fry RC, Begley TJ, Samson LD (2004). «Genome-wide responses to DNA-damaging agents». Annual Review of Microbiology. 59: 357–77. doi:10.1146/annurev.micro.59.031805.133658. PMID 16153173.
- ^ Espejel S, Martín M, Klatt P, Martín-Caballero J, Flores JM, Blasco MA (May 2004). «Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice». EMBO Reports. 5 (5): 503–09. doi:10.1038/sj.embor.7400127. PMC 1299048. PMID 15105825.
- ^ de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, et al. (May 2002). «Premature aging in mice deficient in DNA repair and transcription». Science. 296 (5571): 1276–79. Bibcode:2002Sci…296.1276D. doi:10.1126/science.1070174. PMID 11950998. S2CID 41930529.
- ^ Dollé ME, Busuttil RA, Garcia AM, Wijnhoven S, van Drunen E, Niedernhofer LJ, et al. (April 2006). «Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice». Mutation Research. 596 (1–2): 22–35. doi:10.1016/j.mrfmmm.2005.11.008. PMID 16472827.
- ^ Kobayashi Y, Narumi I, Satoh K, Funayama T, Kikuchi M, Kitayama S, Watanabe H (November 2004). «Radiation response mechanisms of the extremely radioresistant bacterium Deinococcus radiodurans». Uchu Seibutsu Kagaku. 18 (3): 134–35. PMID 15858357.
- ^ Spindler SR (September 2005). «Rapid and reversible induction of the longevity, anticancer and genomic effects of caloric restriction». Mechanisms of Ageing and Development. 126 (9): 960–66. doi:10.1016/j.mad.2005.03.016. PMID 15927235. S2CID 7067036.
- ^ Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, Wu JM, Darzynkiewicz Z (December 2012). «Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage- signaling». Aging. 4 (12): 952–65. doi:10.18632/aging.100521. PMC 3615161. PMID 23363784.
- ^ Tissenbaum HA, Guarente L (March 2001). «Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans». Nature. 410 (6825): 227–30. Bibcode:2001Natur.410..227T. doi:10.1038/35065638. PMID 11242085. S2CID 4356885.
- ^ Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. (July 2004). «Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase». Science. 305 (5682): 390–92. Bibcode:2004Sci…305..390C. doi:10.1126/science.1099196. PMID 15205477. S2CID 33503081.
- ^ Cabelof DC, Yanamadala S, Raffoul JJ, Guo Z, Soofi A, Heydari AR (March 2003). «Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline». DNA Repair. 2 (3): 295–307. doi:10.1016/S1568-7864(02)00219-7. PMID 12547392.
- ^ Stuart JA, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA (March 2004). «Mitochondrial and nuclear DNA base excision repair are affected differently by caloric restriction». FASEB Journal. 18 (3): 595–97. doi:10.1096/fj.03-0890fje. PMID 14734635. S2CID 43118901.
- ^ Walker DW, McColl G, Jenkins NL, Harris J, Lithgow GJ (May 2000). «Evolution of lifespan in C. elegans». Nature. 405 (6784): 296–97. doi:10.1038/35012693. PMID 10830948. S2CID 4402039.
- ^ Johnson G (28 December 2010). «Unearthing Prehistoric Tumors, and Debate». The New York Times.
If we lived long enough, sooner or later we all would get cancer.
- ^ Alberts B, Johnson A, Lewis J, et al. (2002). «The Preventable Causes of Cancer». Molecular biology of the cell (4th ed.). New York: Garland Science. ISBN 978-0-8153-4072-0.
A certain irreducible background incidence of cancer is to be expected regardless of circumstances: mutations can never be absolutely avoided, because they are an inescapable consequence of fundamental limitations on the accuracy of DNA replication, as discussed in Chapter 5. If a human could live long enough, it is inevitable that at least one of his or her cells would eventually accumulate a set of mutations sufficient for cancer to develop.
- ^ Friedenson B (August 2007). «The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers». BMC Cancer. 7: 152. doi:10.1186/1471-2407-7-152. PMC 1959234. PMID 17683622.
- ^ Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, Turchi JJ (April 2016). «DNA repair targeted therapy: The past or future of cancer treatment?». Pharmacology & Therapeutics. 160: 65–83. doi:10.1016/j.pharmthera.2016.02.003. PMC 4811676. PMID 26896565.
- ^ Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. (April 2005). «Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase». Nature. 434 (7035): 913–17. Bibcode:2005Natur.434..913B. doi:10.1038/nature03443. PMID 15829966. S2CID 4391043.
- ^ Goldstein M, Kastan MB (2015). «The DNA damage response: implications for tumor responses to radiation and chemotherapy». Annual Review of Medicine. 66: 129–43. doi:10.1146/annurev-med-081313-121208. PMID 25423595.
- ^ a b Jeggo PA, Pearl LH, Carr AM (January 2016). «DNA repair, genome stability and cancer: a historical perspective» (PDF). Nature Reviews. Cancer. 16 (1): 35–42. doi:10.1038/nrc.2015.4. PMID 26667849. S2CID 14941857.
- ^ Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, et al. (April 2005). «DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis». Nature. 434 (7035): 864–70. Bibcode:2005Natur.434..864B. doi:10.1038/nature03482. PMID 15829956. S2CID 4398393.
- ^ a b Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. (November 2006). «Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints». Nature. 444 (7119): 633–37. Bibcode:2006Natur.444..633B. doi:10.1038/nature05268. PMID 17136093. S2CID 4406956.
- ^ Gaillard H, García-Muse T, Aguilera A (May 2015). «Replication stress and cancer». Nature Reviews. Cancer. 15 (5): 276–89. doi:10.1038/nrc3916. hdl:10261/123721. PMID 25907220. S2CID 11342123.
- ^ Halazonetis TD, Gorgoulis VG, Bartek J (March 2008). «An oncogene-induced DNA damage model for cancer development». Science. 319 (5868): 1352–55. Bibcode:2008Sci…319.1352H. doi:10.1126/science.1140735. PMID 18323444. S2CID 16426080.
- ^ de Boer J, Hoeijmakers JH (March 2000). «Nucleotide excision repair and human syndromes» (PDF). Carcinogenesis. 21 (3): 453–60. doi:10.1093/carcin/21.3.453. PMID 10688865.
- ^ Broustas CG, Lieberman HB (February 2014). «DNA damage response genes and the development of cancer metastasis». Radiation Research. 181 (2): 111–30. Bibcode:2014RadR..181..111B. doi:10.1667/RR13515.1. PMC 4064942. PMID 24397478.
- ^ Zhang P, Wang J, Gao W, Yuan BZ, Rogers J, Reed E (May 2004). «CHK2 kinase expression is down-regulated due to promoter methylation in non-small cell lung cancer». Molecular Cancer. 3 (4): 14. doi:10.1186/1476-4598-3-14. PMC 419366. PMID 15125777.
- ^ Baylin SB, Ohm JE (February 2006). «Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction?». Nature Reviews. Cancer. 6 (2): 107–16. doi:10.1038/nrc1799. PMID 16491070. S2CID 2514545.
- ^ Kanwal R, Gupta S (April 2012). «Epigenetic modifications in cancer». Clinical Genetics. 81 (4): 303–11. doi:10.1111/j.1399-0004.2011.01809.x. PMC 3590802. PMID 22082348.
- ^ Baldassarre G, Battista S, Belletti B, Thakur S, Pentimalli F, Trapasso F, et al. (April 2003). «Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma». Molecular and Cellular Biology. 23 (7): 2225–38. doi:10.1128/MCB.23.7.2225-2238.2003. PMC 150734. PMID 12640109.
- ^ Jacinto FV, Esteller M (July 2007). «Mutator pathways unleashed by epigenetic silencing in human cancer». Mutagenesis. 22 (4): 247–53. doi:10.1093/mutage/gem009. PMID 17412712.
- ^ Lahtz C, Pfeifer GP (February 2011). «Epigenetic changes of DNA repair genes in cancer». Journal of Molecular Cell Biology. 3 (1): 51–58. doi:10.1093/jmcb/mjq053. PMC 3030973. PMID 21278452. Epigenetic changes of DNA repair genes in cancer
- ^ Bernstein C, Nfonsam V, Prasad AR, Bernstein H (March 2013). «Epigenetic field defects in progression to cancer». World Journal of Gastrointestinal Oncology. 5 (3): 43–49. doi:10.4251/wjgo.v5.i3.43. PMC 3648662. PMID 23671730.
- ^ Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (April 1997). «Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2». Proceedings of the National Academy of Sciences of the United States of America. 94 (7): 3122–27. Bibcode:1997PNAS…94.3122N. doi:10.1073/pnas.94.7.3122. PMC 20332. PMID 9096356.
- ^ Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM (December 2006). «Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6». Carcinogenesis. 27 (12): 2402–08. doi:10.1093/carcin/bgl079. PMC 2612936. PMID 16728433.
- ^ Tutt AN, van Oostrom CT, Ross GM, van Steeg H, Ashworth A (March 2002). «Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation». EMBO Reports. 3 (3): 255–60. doi:10.1093/embo-reports/kvf037. PMC 1084010. PMID 11850397.
- ^ German J (March 1969). «Bloom’s syndrome. I. Genetical and clinical observations in the first twenty-seven patients». American Journal of Human Genetics. 21 (2): 196–227. PMC 1706430. PMID 5770175.
- ^ O’Hagan HM, Mohammad HP, Baylin SB (August 2008). «Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island». PLOS Genetics. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- ^ Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, et al. (July 2007). «DNA damage, homology-directed repair, and DNA methylation». PLOS Genetics. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- ^ Malkin D (April 2011). «Li-fraumeni syndrome». Genes & Cancer. 2 (4): 475–84. doi:10.1177/1947601911413466. PMC 3135649. PMID 21779515.
- ^ Fearon ER (November 1997). «Human cancer syndromes: clues to the origin and nature of cancer». Science. 278 (5340): 1043–50. Bibcode:1997Sci…278.1043F. doi:10.1126/science.278.5340.1043. PMID 9353177.
- ^ Halford S, Rowan A, Sawyer E, Talbot I, Tomlinson I (June 2005). «O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G:C>A:T transitions». Gut. 54 (6): 797–802. doi:10.1136/gut.2004.059535. PMC 1774551. PMID 15888787.
- ^ Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, et al. (September 2005). «MGMT promoter methylation and field defect in sporadic colorectal cancer». Journal of the National Cancer Institute. 97 (18): 1330–38. doi:10.1093/jnci/dji275. PMID 16174854.
- ^ Psofaki V, Kalogera C, Tzambouras N, Stephanou D, Tsianos E, Seferiadis K, Kolios G (July 2010). «Promoter methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal adenomas». World Journal of Gastroenterology. 16 (28): 3553–60. doi:10.3748/wjg.v16.i28.3553. PMC 2909555. PMID 20653064.
- ^ Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Park CS, et al. (October 2011). «Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence». Langenbeck’s Archives of Surgery. 396 (7): 1017–26. doi:10.1007/s00423-011-0812-9. PMID 21706233. S2CID 8069716.
- ^ Amatu A, Sartore-Bianchi A, Moutinho C, Belotti A, Bencardino K, Chirico G, et al. (April 2013). «Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer». Clinical Cancer Research. 19 (8): 2265–72. doi:10.1158/1078-0432.CCR-12-3518. PMID 23422094.
- ^ Mokarram P, Zamani M, Kavousipour S, Naghibalhossaini F, Irajie C, Moradi Sarabi M, Hosseini SV (May 2013). «Different patterns of DNA methylation of the two distinct O6-methylguanine-DNA methyltransferase (O6-MGMT) promoter regions in colorectal cancer». Molecular Biology Reports. 40 (5): 3851–57. doi:10.1007/s11033-012-2465-3. PMID 23271133. S2CID 18733871.
- ^ Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, et al. (May 2005). «Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer». Gastroenterology. 128 (5): 1160–71. doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- ^ Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, et al. (April 2010). «Modulation of mismatch repair and genomic stability by miR-155». Proceedings of the National Academy of Sciences of the United States of America. 107 (15): 6982–87. Bibcode:2010PNAS..107.6982V. doi:10.1073/pnas.1002472107. PMC 2872463. PMID 20351277.
- ^ Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, et al. (April 2012). «Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer». Genome Integrity. 3 (1): 3. doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- ^ Human DNA Repair Genes, 15 April 2014, MD Anderson Cancer Center, University of Texas
- ^ Jin B, Robertson KD (2013). «DNA methyltransferases, DNA damage repair, and cancer». Advances in Experimental Medicine and Biology. 754: 3–29. doi:10.1007/978-1-4419-9967-2_1. ISBN 978-1-4419-9966-5. PMC 3707278. PMID 22956494.
- ^ Krishnan K, Steptoe AL, Martin HC, Wani S, Nones K, Waddell N, et al. (February 2013). «MicroRNA-182-5p targets a network of genes involved in DNA repair». RNA. 19 (2): 230–42. doi:10.1261/rna.034926.112. PMC 3543090. PMID 23249749.
- ^ Chaisaingmongkol J, Popanda O, Warta R, Dyckhoff G, Herpel E, Geiselhart L, et al. (December 2012). «Epigenetic screen of human DNA repair genes identifies aberrant promoter methylation of NEIL1 in head and neck squamous cell carcinoma». Oncogene. 31 (49): 5108–16. doi:10.1038/onc.2011.660. PMID 22286769.
- ^ Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, et al. (November 2008). «Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers». Molecular Cancer Research. 6 (11): 1710–17. doi:10.1158/1541-7786.MCR-08-0269. PMC 2948671. PMID 19010819.
- ^ Lam JS, Seligson DB, Yu H, Li A, Eeva M, Pantuck AJ, et al. (August 2006). «Flap endonuclease 1 is overexpressed in prostate cancer and is associated with a high Gleason score». BJU International. 98 (2): 445–51. doi:10.1111/j.1464-410X.2006.06224.x. PMID 16879693. S2CID 22165252.
- ^ Kim JM, Sohn HY, Yoon SY, Oh JH, Yang JO, Kim JH, et al. (January 2005). «Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells». Clinical Cancer Research. 11 (2 Pt 1): 473–82. doi:10.1158/1078-0432.473.11.2. PMID 15701830.
- ^ Wang K, Xie C, Chen D (May 2014). «Flap endonuclease 1 is a promising candidate biomarker in gastric cancer and is involved in cell proliferation and apoptosis». International Journal of Molecular Medicine. 33 (5): 1268–74. doi:10.3892/ijmm.2014.1682. PMID 24590400.
- ^ Krause A, Combaret V, Iacono I, Lacroix B, Compagnon C, Bergeron C, et al. (July 2005). «Genome-wide analysis of gene expression in neuroblastomas detected by mass screening» (PDF). Cancer Letters. 225 (1): 111–20. doi:10.1016/j.canlet.2004.10.035. PMID 15922863. S2CID 44644467.
- ^ Iacobuzio-Donahue CA, Maitra A, Olsen M, Lowe AW, van Heek NT, Rosty C, et al. (April 2003). «Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays». The American Journal of Pathology. 162 (4): 1151–62. doi:10.1016/S0002-9440(10)63911-9. PMC 1851213. PMID 12651607.
- ^ Sato M, Girard L, Sekine I, Sunaga N, Ramirez RD, Kamibayashi C, Minna JD (October 2003). «Increased expression and no mutation of the Flap endonuclease (FEN1) gene in human lung cancer». Oncogene. 22 (46): 7243–46. doi:10.1038/sj.onc.1206977. PMID 14562054.
- ^ Bi FF, Li D, Yang Q (2013). «Hypomethylation of ETS transcription factor binding sites and upregulation of PARP1 expression in endometrial cancer». BioMed Research International. 2013: 946268. doi:10.1155/2013/946268. PMC 3666359. PMID 23762867.
- ^ Bi FF, Li D, Yang Q (February 2013). «Promoter hypomethylation, especially around the E26 transformation-specific motif, and increased expression of poly (ADP-ribose) polymerase 1 in BRCA-mutated serous ovarian cancer». BMC Cancer. 13: 90. doi:10.1186/1471-2407-13-90. PMC 3599366. PMID 23442605.
- ^ Supek F, Lehner B (May 2015). «Differential DNA mismatch repair underlies mutation rate variation across the human genome». Nature. 521 (7550): 81–84. Bibcode:2015Natur.521…81S. doi:10.1038/nature14173. PMC 4425546. PMID 25707793.
- ^ a b Zheng CL, Wang NJ, Chung J, Moslehi H, Sanborn JZ, Hur JS, et al. (November 2014). «Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes». Cell Reports. 9 (4): 1228–34. doi:10.1016/j.celrep.2014.10.031. PMC 4254608. PMID 25456125.
- ^ Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM (April 2013). «The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα». Cell. 153 (3): 590–600. doi:10.1016/j.cell.2013.03.025. PMC 3641580. PMID 23622243.
- ^ Supek F, Lehner B (July 2017). «Clustered Mutation Signatures Reveal that Error-Prone DNA Repair Targets Mutations to Active Genes». Cell. 170 (3): 534–547.e23. doi:10.1016/j.cell.2017.07.003. hdl:10230/35343. PMID 28753428.
- ^ Polak P, Lawrence MS, Haugen E, Stoletzki N, Stojanov P, Thurman RE, et al. (January 2014). «Reduced local mutation density in regulatory DNA of cancer genomes is linked to DNA repair». Nature Biotechnology. 32 (1): 71–75. doi:10.1038/nbt.2778. PMC 4116484. PMID 24336318.
- ^ Swenberg JA, Lu K, Moeller BC, Gao L, Upton PB, Nakamura J, Starr TB (March 2011). «Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment». Toxicol Sci. 120 Suppl 1 (Suppl 1): S130–45. doi:10.1093/toxsci/kfq371. PMC 3043087. PMID 21163908.
- ^ a b Hamilton ML, Guo Z, Fuller CD, Van Remmen H, Ward WF, Austad SN, Troyer DA, Thompson I, Richardson A (May 2001). «A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA». Nucleic Acids Res. 29 (10): 2117–26. doi:10.1093/nar/29.10.2117. PMC 55450. PMID 11353081.
- ^ Ming X, Matter B, Song M, Veliath E, Shanley R, Jones R, Tretyakova N (March 2014). «Mapping structurally defined guanine oxidation products along DNA duplexes: influence of local sequence context and endogenous cytosine methylation». J Am Chem Soc. 136 (11): 4223–35. doi:10.1021/ja411636j. PMC 3985951. PMID 24571128.
- ^ a b c Zhou X, Zhuang Z, Wang W, He L, Wu H, Cao Y, Pan F, Zhao J, Hu Z, Sekhar C, Guo Z (September 2016). «OGG1 is essential in oxidative stress-induced DNA demethylation». Cell Signal. 28 (9): 1163–1171. doi:10.1016/j.cellsig.2016.05.021. PMID 27251462.
- ^ Poetsch AR (2020). «The genomics of oxidative DNA damage, repair, and resulting mutagenesis». Comput Struct Biotechnol J. 18: 207–219. doi:10.1016/j.csbj.2019.12.013. PMC 6974700. PMID 31993111.
- ^ D’Augustin O, Huet S, Campalans A, Radicella JP (November 2020). «Lost in the Crowd: How Does Human 8-Oxoguanine DNA Glycosylase 1 (OGG1) Find 8-Oxoguanine in the Genome?». Int J Mol Sci. 21 (21): 8360. doi:10.3390/ijms21218360. PMC 7664663. PMID 33171795.
- ^ Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A (September 2004). «In situ analysis of repair processes for oxidative DNA damage in mammalian cells». Proc Natl Acad Sci U S A. 101 (38): 13738–43. Bibcode:2004PNAS..10113738L. doi:10.1073/pnas.0406048101. PMC 518826. PMID 15365186.
- ^ Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, Costello JF, Wilkinson MF, Joung JK (December 2013). «Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins». Nat. Biotechnol. 31 (12): 1137–42. doi:10.1038/nbt.2726. PMC 3858462. PMID 24108092.
- ^ Ding N, Bonham EM, Hannon BE, Amick TR, Baylin SB, O’Hagan HM (June 2016). «Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage». J Mol Cell Biol. 8 (3): 244–54. doi:10.1093/jmcb/mjv050. PMC 4937888. PMID 26186941.
- ^ a b Jiang Z, Lai Y, Beaver JM, Tsegay PS, Zhao ML, Horton JK, Zamora M, Rein HL, Miralles F, Shaver M, Hutcheson JD, Agoulnik I, Wilson SH, Liu Y (January 2020). «Oxidative DNA Damage Modulates DNA Methylation Pattern in Human Breast Cancer 1 (BRCA1) Gene via the Crosstalk between DNA Polymerase β and a de novo DNA Methyltransferase». Cells. 9 (1): 225. doi:10.3390/cells9010225. PMC 7016758. PMID 31963223.
- ^ Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H (June 2005). «Recruitment of DNA methyltransferase I to DNA repair sites». Proc Natl Acad Sci U S A. 102 (25): 8905–9. Bibcode:2005PNAS..102.8905M. doi:10.1073/pnas.0501034102. PMC 1157029. PMID 15956212.
- ^ a b Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, Messina S, Iuliano R, Fusco A, Santillo MR, Muller MT, Chiariotti L, Gottesman ME, Avvedimento EV (July 2007). «DNA damage, homology-directed repair, and DNA methylation». PLOS Genet. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- ^ a b O’Hagan HM, Mohammad HP, Baylin SB (August 2008). «Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island». PLOS Genet. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- ^ Ha K, Lee GE, Palii SS, Brown KD, Takeda Y, Liu K, Bhalla KN, Robertson KD (January 2011). «Rapid and transient recruitment of DNMT1 to DNA double-strand breaks is mediated by its interaction with multiple components of the DNA damage response machinery». Hum Mol Genet. 20 (1): 126–40. doi:10.1093/hmg/ddq451. PMC 3000680. PMID 20940144.
- ^ Russo G, Landi R, Pezone A, Morano A, Zuchegna C, Romano A, Muller MT, Gottesman ME, Porcellini A, Avvedimento EV (September 2016). «DNA damage and Repair Modify DNA methylation and Chromatin Domain of the Targeted Locus: Mechanism of allele methylation polymorphism». Sci Rep. 6: 33222. Bibcode:2016NatSR…633222R. doi:10.1038/srep33222. PMC 5024116. PMID 27629060.
- ^ Farris MH, Texter PA, Mora AA, Wiles MV, Mac Garrigle EF, Klaus SA, Rosfjord K (December 2020). «Detection of CRISPR-mediated genome modifications through altered methylation patterns of CpG islands». BMC Genomics. 21 (1): 856. doi:10.1186/s12864-020-07233-2. PMC 7709351. PMID 33267773.
- ^ Allen B, Pezone A, Porcellini A, Muller MT, Masternak MM (June 2017). «Non-homologous end joining induced alterations in DNA methylation: A source of permanent epigenetic change». Oncotarget. 8 (25): 40359–40372. doi:10.18632/oncotarget.16122. PMC 5522286. PMID 28423717.
- ^ Cromie GA, Connelly JC, Leach DR (December 2001). «Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans». Molecular Cell. 8 (6): 1163–74. doi:10.1016/S1097-2765(01)00419-1. PMID 11779493.
- ^ O’Brien PJ (February 2006). «Catalytic promiscuity and the divergent evolution of DNA repair enzymes». Chemical Reviews. 106 (2): 720–52. doi:10.1021/cr040481v. PMID 16464022.
- ^ Maresca B, Schwartz JH (January 2006). «Sudden origins: a general mechanism of evolution based on stress protein concentration and rapid environmental change». The Anatomical Record Part B: The New Anatomist. 289 (1): 38–46. doi:10.1002/ar.b.20089. PMID 16437551.
- ^ «CRISPR gene-editing tool has scientists thrilled – but nervous» CBC news. Author Kelly Crowe. 30 November 2015.
External links[edit]
![]()
This audio file was created from a revision of this article dated 17 June 2005, and does not reflect subsequent edits.
 Media related to DNA repair at Wikimedia Commons
Media related to DNA repair at Wikimedia Commons- Roswell Park Cancer Institute DNA Repair Lectures
- A comprehensive list of Human DNA Repair Genes
- 3D structures of some DNA repair enzymes
- Human DNA repair diseases
- DNA repair special interest group
- DNA Repair Archived 12 February 2018 at the Wayback Machine
- DNA Damage and DNA Repair
- Segmental Progeria
- DNA-damage repair; the good, the bad, and the ugly
DNA damage resulting in multiple broken chromosomes
DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome.[1] In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day.[2] Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell’s ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell’s genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur, including double-strand breaks and DNA crosslinkages (interstrand crosslinks or ICLs).[3][4] This can eventually lead to malignant tumors, or cancer as per the two hit hypothesis.
The rate of DNA repair is dependent on many factors, including the cell type, the age of the cell, and the extracellular environment. A cell that has accumulated a large amount of DNA damage, or one that no longer effectively repairs damage incurred to its DNA, can enter one of three possible states:
- an irreversible state of dormancy, known as senescence
- cell suicide, also known as apoptosis or programmed cell death
- unregulated cell division, which can lead to the formation of a tumor that is cancerous
The DNA repair ability of a cell is vital to the integrity of its genome and thus to the normal functionality of that organism. Many genes that were initially shown to influence life span have turned out to be involved in DNA damage repair and protection.[5]
Paul Modrich talks about himself and his work in DNA repair.
The 2015 Nobel Prize in Chemistry was awarded to Tomas Lindahl, Paul Modrich, and Aziz Sancar for their work on the molecular mechanisms of DNA repair processes.[6][7]
DNA damage[edit]
DNA damage, due to environmental factors and normal metabolic processes inside the cell, occurs at a rate of 10,000 to 1,000,000 molecular lesions per cell per day.[2] While this constitutes only 0.000165% of the human genome’s approximately 6 billion bases, unrepaired lesions in critical genes (such as tumor suppressor genes) can impede a cell’s ability to carry out its function and appreciably increase the likelihood of tumor formation and contribute to tumour heterogeneity.
The vast majority of DNA damage affects the primary structure of the double helix; that is, the bases themselves are chemically modified. These modifications can in turn disrupt the molecules’ regular helical structure by introducing non-native chemical bonds or bulky adducts that do not fit in the standard double helix. Unlike proteins and RNA, DNA usually lacks tertiary structure and therefore damage or disturbance does not occur at that level. DNA is, however, supercoiled and wound around «packaging» proteins called histones (in eukaryotes), and both superstructures are vulnerable to the effects of DNA damage.
Sources[edit]
DNA damage can be subdivided into two main types:
- endogenous damage such as attack by reactive oxygen species produced from normal metabolic byproducts (spontaneous mutation), especially the process of oxidative deamination
- also includes replication errors
- exogenous damage caused by external agents such as
- ultraviolet [UV 200–400 nm] radiation from the sun or other artificial light sources
- other radiation frequencies, including x-rays and gamma rays
- hydrolysis or thermal disruption
- certain plant toxins
- human-made mutagenic chemicals, especially aromatic compounds that act as DNA intercalating agents
- viruses[8]
The replication of damaged DNA before cell division can lead to the incorporation of wrong bases opposite damaged ones. Daughter cells that inherit these wrong bases carry mutations from which the original DNA sequence is unrecoverable (except in the rare case of a back mutation, for example, through gene conversion).
Types[edit]
There are several types of damage to DNA due to endogenous cellular processes:
- oxidation of bases [e.g. 8-oxo-7,8-dihydroguanine (8-oxoG)] and generation of DNA strand interruptions from reactive oxygen species,
- alkylation of bases (usually methylation), such as formation of 7-methylguanosine, 1-methyladenine, 6-O-Methylguanine
- hydrolysis of bases, such as deamination, depurination, and depyrimidination.
- «bulky adduct formation» (e.g., benzo[a]pyrene diol epoxide-dG adduct, aristolactam I-dA adduct)
- mismatch of bases, due to errors in DNA replication, in which the wrong DNA base is stitched into place in a newly forming DNA strand, or a DNA base is skipped over or mistakenly inserted.
- Monoadduct damage cause by change in single nitrogenous base of DNA
- Diadduct damage
Damage caused by exogenous agents comes in many forms. Some examples are:
- UV-B light causes crosslinking between adjacent cytosine and thymine bases creating pyrimidine dimers. This is called direct DNA damage.
- UV-A light creates mostly free radicals. The damage caused by free radicals is called indirect DNA damage.
- Ionizing radiation such as that created by radioactive decay or in cosmic rays causes breaks in DNA strands. Intermediate-level ionizing radiation may induce irreparable DNA damage (leading to replicational and transcriptional errors needed for neoplasia or may trigger viral interactions) leading to pre-mature aging and cancer.
- Thermal disruption at elevated temperature increases the rate of depurination (loss of purine bases from the DNA backbone) and single-strand breaks. For example, hydrolytic depurination is seen in the thermophilic bacteria, which grow in hot springs at 40–80 °C.[9][10] The rate of depurination (300 purine residues per genome per generation) is too high in these species to be repaired by normal repair machinery, hence a possibility of an adaptive response cannot be ruled out.
- Industrial chemicals such as vinyl chloride and hydrogen peroxide, and environmental chemicals such as polycyclic aromatic hydrocarbons found in smoke, soot and tar create a huge diversity of DNA adducts- ethenobases, oxidized bases, alkylated phosphotriesters and crosslinking of DNA, just to name a few.
UV damage, alkylation/methylation, X-ray damage and oxidative damage are examples of induced damage. Spontaneous damage can include the loss of a base, deamination, sugar ring puckering and tautomeric shift. Constitutive (spontaneous) DNA damage caused by endogenous oxidants can be detected as a low level of histone H2AX phosphorylation in untreated cells.[11]
Nuclear versus mitochondrial[edit]
In human cells, and eukaryotic cells in general, DNA is found in two cellular locations – inside the nucleus and inside the mitochondria. Nuclear DNA (nDNA) exists as chromatin during non-replicative stages of the cell cycle and is condensed into aggregate structures known as chromosomes during cell division. In either state the DNA is highly compacted and wound up around bead-like proteins called histones. Whenever a cell needs to express the genetic information encoded in its nDNA the required chromosomal region is unravelled, genes located therein are expressed, and then the region is condensed back to its resting conformation. Mitochondrial DNA (mtDNA) is located inside mitochondria organelles, exists in multiple copies, and is also tightly associated with a number of proteins to form a complex known as the nucleoid. Inside mitochondria, reactive oxygen species (ROS), or free radicals, byproducts of the constant production of adenosine triphosphate (ATP) via oxidative phosphorylation, create a highly oxidative environment that is known to damage mtDNA. A critical enzyme in counteracting the toxicity of these species is superoxide dismutase, which is present in both the mitochondria and cytoplasm of eukaryotic cells.
Senescence and apoptosis[edit]
Senescence, an irreversible process in which the cell no longer divides, is a protective response to the shortening of the chromosome ends, called telomeres. The telomeres are long regions of repetitive noncoding DNA that cap chromosomes and undergo partial degradation each time a cell undergoes division (see Hayflick limit).[12] In contrast, quiescence is a reversible state of cellular dormancy that is unrelated to genome damage (see cell cycle). Senescence in cells may serve as a functional alternative to apoptosis in cases where the physical presence of a cell for spatial reasons is required by the organism,[13] which serves as a «last resort» mechanism to prevent a cell with damaged DNA from replicating inappropriately in the absence of pro-growth cellular signaling. Unregulated cell division can lead to the formation of a tumor (see cancer), which is potentially lethal to an organism. Therefore, the induction of senescence and apoptosis is considered to be part of a strategy of protection against cancer.[14]
Mutation[edit]
It is important to distinguish between DNA damage and mutation, the two major types of error in DNA. DNA damage and mutation are fundamentally different. Damage results in physical abnormalities in the DNA, such as single- and double-strand breaks, 8-hydroxydeoxyguanosine residues, and polycyclic aromatic hydrocarbon adducts. DNA damage can be recognized by enzymes, and thus can be correctly repaired if redundant information, such as the undamaged sequence in the complementary DNA strand or in a homologous chromosome, is available for copying. If a cell retains DNA damage, transcription of a gene can be prevented, and thus translation into a protein will also be blocked. Replication may also be blocked or the cell may die.
In contrast to DNA damage, a mutation is a change in the base sequence of the DNA. A mutation cannot be recognized by enzymes once the base change is present in both DNA strands, and thus a mutation cannot be repaired. At the cellular level, mutations can cause alterations in protein function and regulation. Mutations are replicated when the cell replicates. In a population of cells, mutant cells will increase or decrease in frequency according to the effects of the mutation on the ability of the cell to survive and reproduce.
Although distinctly different from each other, DNA damage and mutation are related because DNA damage often causes errors of DNA synthesis during replication or repair; these errors are a major source of mutation.
Given these properties of DNA damage and mutation, it can be seen that DNA damage is a special problem in non-dividing or slowly-dividing cells, where unrepaired damage will tend to accumulate over time. On the other hand, in rapidly dividing cells, unrepaired DNA damage that does not kill the cell by blocking replication will tend to cause replication errors and thus mutation. The great majority of mutations that are not neutral in their effect are deleterious to a cell’s survival. Thus, in a population of cells composing a tissue with replicating cells, mutant cells will tend to be lost. However, infrequent mutations that provide a survival advantage will tend to clonally expand at the expense of neighboring cells in the tissue. This advantage to the cell is disadvantageous to the whole organism because such mutant cells can give rise to cancer. Thus, DNA damage in frequently dividing cells, because it gives rise to mutations, is a prominent cause of cancer. In contrast, DNA damage in infrequently-dividing cells is likely a prominent cause of aging.[15]
Mechanisms[edit]
Cells cannot function if DNA damage corrupts the integrity and accessibility of essential information in the genome (but cells remain superficially functional when non-essential genes are missing or damaged). Depending on the type of damage inflicted on the DNA’s double helical structure, a variety of repair strategies have evolved to restore lost information. If possible, cells use the unmodified complementary strand of the DNA or the sister chromatid as a template to recover the original information. Without access to a template, cells use an error-prone recovery mechanism known as translesion synthesis as a last resort.
Damage to DNA alters the spatial configuration of the helix, and such alterations can be detected by the cell. Once damage is localized, specific DNA repair molecules bind at or near the site of damage, inducing other molecules to bind and form a complex that enables the actual repair to take place.
Direct reversal[edit]
Cells are known to eliminate three types of damage to their DNA by chemically reversing it. These mechanisms do not require a template, since the types of damage they counteract can occur in only one of the four bases. Such direct reversal mechanisms are specific to the type of damage incurred and do not involve breakage of the phosphodiester backbone. The formation of pyrimidine dimers upon irradiation with UV light results in an abnormal covalent bond between adjacent pyrimidine bases. The photoreactivation process directly reverses this damage by the action of the enzyme photolyase, whose activation is obligately dependent on energy absorbed from blue/UV light (300–500 nm wavelength) to promote catalysis.[16] Photolyase, an old enzyme present in bacteria, fungi, and most animals no longer functions in humans,[17] who instead use nucleotide excision repair to repair damage from UV irradiation. Another type of damage, methylation of guanine bases, is directly reversed by the enzyme methyl guanine methyl transferase (MGMT), the bacterial equivalent of which is called ogt. This is an expensive process because each MGMT molecule can be used only once; that is, the reaction is stoichiometric rather than catalytic.[18] A generalized response to methylating agents in bacteria is known as the adaptive response and confers a level of resistance to alkylating agents upon sustained exposure by upregulation of alkylation repair enzymes.[19] The third type of DNA damage reversed by cells is certain methylation of the bases cytosine and adenine.
Single-strand damage[edit]
Structure of the base-excision repair enzyme uracil-DNA glycosylase excising a hydrolytically-produced uracil residue from DNA. The uracil residue is shown in yellow.
When only one of the two strands of a double helix has a defect, the other strand can be used as a template to guide the correction of the damaged strand. In order to repair damage to one of the two paired molecules of DNA, there exist a number of excision repair mechanisms that remove the damaged nucleotide and replace it with an undamaged nucleotide complementary to that found in the undamaged DNA strand.[18]
- Base excision repair (BER): damaged single bases or nucleotides are most commonly repaired by removing the base or the nucleotide involved and then inserting the correct base or nucleotide. In base excision repair, a glycosylase[20] enzyme removes the damaged base from the DNA by cleaving the bond between the base and the deoxyribose. These enzymes remove a single base to create an apurinic or apyrimidinic site (AP site).[20] Enzymes called AP endonucleases nick the damaged DNA backbone at the AP site. DNA polymerase then removes the damaged region using its 5’ to 3’ exonuclease activity and correctly synthesizes the new strand using the complementary strand as a template.[20] The gap is then sealed by enzyme DNA ligase.[21]
- Nucleotide excision repair (NER): bulky, helix-distorting damage, such as pyrimidine dimerization caused by UV light is usually repaired by a three-step process. First the damage is recognized, then 12-24 nucleotide-long strands of DNA are removed both upstream and downstream of the damage site by endonucleases, and the removed DNA region is then resynthesized.[22] NER is a highly evolutionarily conserved repair mechanism and is used in nearly all eukaryotic and prokaryotic cells.[22] In prokaryotes, NER is mediated by Uvr proteins.[22] In eukaryotes, many more proteins are involved, although the general strategy is the same.[22]
- Mismatch repair systems are present in essentially all cells to correct errors that are not corrected by proofreading. These systems consist of at least two proteins. One detects the mismatch, and the other recruits an endonuclease that cleaves the newly synthesized DNA strand close to the region of damage. In E. coli , the proteins involved are the Mut class proteins: MutS, MutL, and MutH. In most Eukaryotes, the analog for MutS is MSH and the analog for MutL is MLH. MutH is only present in bacteria. This is followed by removal of damaged region by an exonuclease, resynthesis by DNA polymerase, and nick sealing by DNA ligase.[23]
Double-strand breaks[edit]
The main double-strand break repair pathways
Double-strand breaks, in which both strands in the double helix are severed, are particularly hazardous to the cell because they can lead to genome rearrangements. In fact, when a double-strand break is accompanied by a cross-linkage joining the two strands at the same point, neither strand can be used as a template for the repair mechanisms, so that the cell will not be able to complete mitosis when it next divides, and will either die or, in rare cases, undergo a mutation.[3][4] Three mechanisms exist to repair double-strand breaks (DSBs): non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homologous recombination (HR):[18][24]
DNA ligase, shown above repairing chromosomal damage, is an enzyme that joins broken nucleotides together by catalyzing the formation of an internucleotide ester bond between the phosphate backbone and the deoxyribose nucleotides.
- In NHEJ, DNA Ligase IV, a specialized DNA ligase that forms a complex with the cofactor XRCC4, directly joins the two ends.[25] To guide accurate repair, NHEJ relies on short homologous sequences called microhomologies present on the single-stranded tails of the DNA ends to be joined. If these overhangs are compatible, repair is usually accurate.[26][27][28][29] NHEJ can also introduce mutations during repair. Loss of damaged nucleotides at the break site can lead to deletions, and joining of nonmatching termini forms insertions or translocations. NHEJ is especially important before the cell has replicated its DNA, since there is no template available for repair by homologous recombination. There are «backup» NHEJ pathways in higher eukaryotes.[30] Besides its role as a genome caretaker, NHEJ is required for joining hairpin-capped double-strand breaks induced during V(D)J recombination, the process that generates diversity in B-cell and T-cell receptors in the vertebrate immune system.[31]
- MMEJ starts with short-range end resection by MRE11 nuclease on either side of a double-strand break to reveal microhomology regions.[32] In further steps,[33] Poly (ADP-ribose) polymerase 1 (PARP1) is required and may be an early step in MMEJ. There is pairing of microhomology regions followed by recruitment of flap structure-specific endonuclease 1 (FEN1) to remove overhanging flaps. This is followed by recruitment of XRCC1–LIG3 to the site for ligating the DNA ends, leading to an intact DNA. MMEJ is always accompanied by a deletion, so that MMEJ is a mutagenic pathway for DNA repair.[34]
- HR requires the presence of an identical or nearly identical sequence to be used as a template for repair of the break. The enzymatic machinery responsible for this repair process is nearly identical to the machinery responsible for chromosomal crossover during meiosis. This pathway allows a damaged chromosome to be repaired using a sister chromatid (available in G2 after DNA replication) or a homologous chromosome as a template. DSBs caused by the replication machinery attempting to synthesize across a single-strand break or unrepaired lesion cause collapse of the replication fork and are typically repaired by recombination.
In an in vitro system, MMEJ occurred in mammalian cells at the levels of 10–20% of HR when both HR and NHEJ mechanisms were also available.[32]
The extremophile Deinococcus radiodurans has a remarkable ability to survive DNA damage from ionizing radiation and other sources. At least two copies of the genome, with random DNA breaks, can form DNA fragments through annealing. Partially overlapping fragments are then used for synthesis of homologous regions through a moving D-loop that can continue extension until complementary partner strands are found. In the final step, there is crossover by means of RecA-dependent homologous recombination.[35]
Topoisomerases introduce both single- and double-strand breaks in the course of changing the DNA’s state of supercoiling, which is especially common in regions near an open replication fork. Such breaks are not considered DNA damage because they are a natural intermediate in the topoisomerase biochemical mechanism and are immediately repaired by the enzymes that created them.
Another type of DNA double-strand breaks originates from the DNA heat-sensitive or heat-labile sites. These DNA sites are not initial DSBs. However, they convert to DSB after treating with elevated temperature. Ionizing irradiation can induces a highly complex form of DNA damage as clustered damage. It consists of different types of DNA lesions in various locations of the DNA helix. Some of these closely located lesions can probably convert to DSB by exposure to high temperatures. But the exact nature of these lesions and their interactions is not yet known[36]
Translesion synthesis[edit]
Translesion synthesis (TLS) is a DNA damage tolerance process that allows the DNA replication machinery to replicate past DNA lesions such as thymine dimers or AP sites.[37] It involves switching out regular DNA polymerases for specialized translesion polymerases (i.e. DNA polymerase IV or V, from the Y Polymerase family), often with larger active sites that can facilitate the insertion of bases opposite damaged nucleotides. The polymerase switching is thought to be mediated by, among other factors, the post-translational modification of the replication processivity factor PCNA. Translesion synthesis polymerases often have low fidelity (high propensity to insert wrong bases) on undamaged templates relative to regular polymerases. However, many are extremely efficient at inserting correct bases opposite specific types of damage. For example, Pol η mediates error-free bypass of lesions induced by UV irradiation, whereas Pol ι introduces mutations at these sites. Pol η is known to add the first adenine across the T^T photodimer using Watson-Crick base pairing and the second adenine will be added in its syn conformation using Hoogsteen base pairing. From a cellular perspective, risking the introduction of point mutations during translesion synthesis may be preferable to resorting to more drastic mechanisms of DNA repair, which may cause gross chromosomal aberrations or cell death. In short, the process involves specialized polymerases either bypassing or repairing lesions at locations of stalled DNA replication. For example, Human DNA polymerase eta can bypass complex DNA lesions like guanine-thymine intra-strand crosslink, G[8,5-Me]T, although it can cause targeted and semi-targeted mutations.[38] Paromita Raychaudhury and Ashis Basu[39] studied the toxicity and mutagenesis of the same lesion in Escherichia coli by replicating a G[8,5-Me]T-modified plasmid in E. coli with specific DNA polymerase knockouts. Viability was very low in a strain lacking pol II, pol IV, and pol V, the three SOS-inducible DNA polymerases, indicating that translesion synthesis is conducted primarily by these specialized DNA polymerases.
A bypass platform is provided to these polymerases by Proliferating cell nuclear antigen (PCNA). Under normal circumstances, PCNA bound to polymerases replicates the DNA. At a site of lesion, PCNA is ubiquitinated, or modified, by the RAD6/RAD18 proteins to provide a platform for the specialized polymerases to bypass the lesion and resume DNA replication.[40][41] After translesion synthesis, extension is required. This extension can be carried out by a replicative polymerase if the TLS is error-free, as in the case of Pol η, yet if TLS results in a mismatch, a specialized polymerase is needed to extend it; Pol ζ. Pol ζ is unique in that it can extend terminal mismatches, whereas more processive polymerases cannot. So when a lesion is encountered, the replication fork will stall, PCNA will switch from a processive polymerase to a TLS polymerase such as Pol ι to fix the lesion, then PCNA may switch to Pol ζ to extend the mismatch, and last PCNA will switch to the processive polymerase to continue replication.
Global response to DNA damage[edit]
Cells exposed to ionizing radiation, ultraviolet light or chemicals are prone to acquire multiple sites of bulky DNA lesions and double-strand breaks. Moreover, DNA damaging agents can damage other biomolecules such as proteins, carbohydrates, lipids, and RNA. The accumulation of damage, to be specific, double-strand breaks or adducts stalling the replication forks, are among known stimulation signals for a global response to DNA damage.[42] The global response to damage is an act directed toward the cells’ own preservation and triggers multiple pathways of macromolecular repair, lesion bypass, tolerance, or apoptosis. The common features of global response are induction of multiple genes, cell cycle arrest, and inhibition of cell division.
Initial steps[edit]
The packaging of eukaryotic DNA into chromatin presents a barrier to all DNA-based processes that require recruitment of enzymes to their sites of action. To allow DNA repair, the chromatin must be remodeled. In eukaryotes, ATP dependent chromatin remodeling complexes and histone-modifying enzymes are two predominant factors employed to accomplish this remodeling process.[43]
Chromatin relaxation occurs rapidly at the site of a DNA damage.[44][45] In one of the earliest steps, the stress-activated protein kinase, c-Jun N-terminal kinase (JNK), phosphorylates SIRT6 on serine 10 in response to double-strand breaks or other DNA damage.[46] This post-translational modification facilitates the mobilization of SIRT6 to DNA damage sites, and is required for efficient recruitment of poly (ADP-ribose) polymerase 1 (PARP1) to DNA break sites and for efficient repair of DSBs.[46] PARP1 protein starts to appear at DNA damage sites in less than a second, with half maximum accumulation within 1.6 seconds after the damage occurs.[47] PARP1 synthesizes polymeric adenosine diphosphate ribose (poly (ADP-ribose) or PAR) chains on itself. Next the chromatin remodeler ALC1 quickly attaches to the product of PARP1 action, a poly-ADP ribose chain, and ALC1 completes arrival at the DNA damage within 10 seconds of the occurrence of the damage.[45] About half of the maximum chromatin relaxation, presumably due to action of ALC1, occurs by 10 seconds.[45] This then allows recruitment of the DNA repair enzyme MRE11, to initiate DNA repair, within 13 seconds.[47]
γH2AX, the phosphorylated form of H2AX is also involved in the early steps leading to chromatin decondensation after DNA double-strand breaks. The histone variant H2AX constitutes about 10% of the H2A histones in human chromatin.[48] γH2AX (H2AX phosphorylated on serine 139) can be detected as soon as 20 seconds after irradiation of cells (with DNA double-strand break formation), and half maximum accumulation of γH2AX occurs in one minute.[48] The extent of chromatin with phosphorylated γH2AX is about two million base pairs at the site of a DNA double-strand break.[48] γH2AX does not, itself, cause chromatin decondensation, but within 30 seconds of irradiation, RNF8 protein can be detected in association with γH2AX.[49] RNF8 mediates extensive chromatin decondensation, through its subsequent interaction with CHD4,[50] a component of the nucleosome remodeling and deacetylase complex NuRD.
DDB2 occurs in a heterodimeric complex with DDB1. This complex further complexes with the ubiquitin ligase protein CUL4A[51] and with PARP1.[52] This larger complex rapidly associates with UV-induced damage within chromatin, with half-maximum association completed in 40 seconds.[51] The PARP1 protein, attached to both DDB1 and DDB2, then PARylates (creates a poly-ADP ribose chain) on DDB2 that attracts the DNA remodeling protein ALC1.[52] Action of ALC1 relaxes the chromatin at the site of UV damage to DNA. This relaxation allows other proteins in the nucleotide excision repair pathway to enter the chromatin and repair UV-induced cyclobutane pyrimidine dimer damages.
After rapid chromatin remodeling, cell cycle checkpoints are activated to allow DNA repair to occur before the cell cycle progresses. First, two kinases, ATM and ATR are activated within 5 or 6 minutes after DNA is damaged. This is followed by phosphorylation of the cell cycle checkpoint protein Chk1, initiating its function, about 10 minutes after DNA is damaged.[53]
DNA damage checkpoints[edit]
After DNA damage, cell cycle checkpoints are activated. Checkpoint activation pauses the cell cycle and gives the cell time to repair the damage before continuing to divide. DNA damage checkpoints occur at the G1/S and G2/M boundaries. An intra-S checkpoint also exists. Checkpoint activation is controlled by two master kinases, ATM and ATR. ATM responds to DNA double-strand breaks and disruptions in chromatin structure,[54] whereas ATR primarily responds to stalled replication forks. These kinases phosphorylate downstream targets in a signal transduction cascade, eventually leading to cell cycle arrest. A class of checkpoint mediator proteins including BRCA1, MDC1, and 53BP1 has also been identified.[55] These proteins seem to be required for transmitting the checkpoint activation signal to downstream proteins.
DNA damage checkpoint is a signal transduction pathway that blocks cell cycle progression in G1, G2 and metaphase and slows down the rate of S phase progression when DNA is damaged. It leads to a pause in cell cycle allowing the cell time to repair the damage before continuing to divide.
Checkpoint Proteins can be separated into four groups: phosphatidylinositol 3-kinase (PI3K)-like protein kinase, proliferating cell nuclear antigen (PCNA)-like group, two serine/threonine(S/T) kinases and their adaptors. Central to all DNA damage induced checkpoints responses is a pair of large protein kinases belonging to the first group of PI3K-like protein kinases-the ATM (Ataxia telangiectasia mutated) and ATR (Ataxia- and Rad-related) kinases, whose sequence and functions have been well conserved in evolution. All DNA damage response requires either ATM or ATR because they have the ability to bind to the chromosomes at the site of DNA damage, together with accessory proteins that are platforms on which DNA damage response components and DNA repair complexes can be assembled.
An important downstream target of ATM and ATR is p53, as it is required for inducing apoptosis following DNA damage.[56] The cyclin-dependent kinase inhibitor p21 is induced by both p53-dependent and p53-independent mechanisms and can arrest the cell cycle at the G1/S and G2/M checkpoints by deactivating cyclin/cyclin-dependent kinase complexes.[57]
The prokaryotic SOS response[edit]
The SOS response is the changes in gene expression in Escherichia coli and other bacteria in response to extensive DNA damage. The prokaryotic SOS system is regulated by two key proteins: LexA and RecA. The LexA homodimer is a transcriptional repressor that binds to operator sequences commonly referred to as SOS boxes. In Escherichia coli it is known that LexA regulates transcription of approximately 48 genes including the lexA and recA genes.[58] The SOS response is known to be widespread in the Bacteria domain, but it is mostly absent in some bacterial phyla, like the Spirochetes.[59]
The most common cellular signals activating the SOS response are regions of single-stranded DNA (ssDNA), arising from stalled replication forks or double-strand breaks, which are processed by DNA helicase to separate the two DNA strands.[42] In the initiation step, RecA protein binds to ssDNA in an ATP hydrolysis driven reaction creating RecA–ssDNA filaments. RecA–ssDNA filaments activate LexA autoprotease activity, which ultimately leads to cleavage of LexA dimer and subsequent LexA degradation. The loss of LexA repressor induces transcription of the SOS genes and allows for further signal induction, inhibition of cell division and an increase in levels of proteins responsible for damage processing.
In Escherichia coli, SOS boxes are 20-nucleotide long sequences near promoters with palindromic structure and a high degree of sequence conservation. In other classes and phyla, the sequence of SOS boxes varies considerably, with different length and composition, but it is always highly conserved and one of the strongest short signals in the genome.[59] The high information content of SOS boxes permits differential binding of LexA to different promoters and allows for timing of the SOS response. The lesion repair genes are induced at the beginning of SOS response. The error-prone translesion polymerases, for example, UmuCD’2 (also called DNA polymerase V), are induced later on as a last resort.[60] Once the DNA damage is repaired or bypassed using polymerases or through recombination, the amount of single-stranded DNA in cells is decreased, lowering the amounts of RecA filaments decreases cleavage activity of LexA homodimer, which then binds to the SOS boxes near promoters and restores normal gene expression.
Eukaryotic transcriptional responses to DNA damage[edit]
Eukaryotic cells exposed to DNA damaging agents also activate important defensive pathways by inducing multiple proteins involved in DNA repair, cell cycle checkpoint control, protein trafficking and degradation. Such genome wide transcriptional response is very complex and tightly regulated, thus allowing coordinated global response to damage. Exposure of yeast Saccharomyces cerevisiae to DNA damaging agents results in overlapping but distinct transcriptional profiles. Similarities to environmental shock response indicates that a general global stress response pathway exist at the level of transcriptional activation. In contrast, different human cell types respond to damage differently indicating an absence of a common global response. The probable explanation for this difference between yeast and human cells may be in the heterogeneity of mammalian cells. In an animal different types of cells are distributed among different organs that have evolved different sensitivities to DNA damage.[61]
In general global response to DNA damage involves expression of multiple genes responsible for postreplication repair, homologous recombination, nucleotide excision repair, DNA damage checkpoint, global transcriptional activation, genes controlling mRNA decay, and many others. A large amount of damage to a cell leaves it with an important decision: undergo apoptosis and die, or survive at the cost of living with a modified genome. An increase in tolerance to damage can lead to an increased rate of survival that will allow a greater accumulation of mutations. Yeast Rev1 and human polymerase η are members of Y family translesion DNA polymerases present during global response to DNA damage and are responsible for enhanced mutagenesis during a global response to DNA damage in eukaryotes.[42]
Aging[edit]
Pathological effects of poor DNA repair[edit]
DNA repair rate is an important determinant of cell pathology.
Experimental animals with genetic deficiencies in DNA repair often show decreased life span and increased cancer incidence.[15] For example, mice deficient in the dominant NHEJ pathway and in telomere maintenance mechanisms get lymphoma and infections more often, and, as a consequence, have shorter lifespans than wild-type mice.[62] In similar manner, mice deficient in a key repair and transcription protein that unwinds DNA helices have premature onset of aging-related diseases and consequent shortening of lifespan.[63] However, not every DNA repair deficiency creates exactly the predicted effects; mice deficient in the NER pathway exhibited shortened life span without correspondingly higher rates of mutation.[64]
If the rate of DNA damage exceeds the capacity of the cell to repair it, the accumulation of errors can overwhelm the cell and result in early senescence, apoptosis, or cancer. Inherited diseases associated with faulty DNA repair functioning result in premature aging,[15] increased sensitivity to carcinogens and correspondingly increased cancer risk (see below). On the other hand, organisms with enhanced DNA repair systems, such as Deinococcus radiodurans, the most radiation-resistant known organism, exhibit remarkable resistance to the double-strand break-inducing effects of radioactivity, likely due to enhanced efficiency of DNA repair and especially NHEJ.[65]
Longevity and caloric restriction[edit]
Most life span influencing genes affect the rate of DNA damage.
A number of individual genes have been identified as influencing variations in life span within a population of organisms. The effects of these genes is strongly dependent on the environment, in particular, on the organism’s diet. Caloric restriction reproducibly results in extended lifespan in a variety of organisms, likely via nutrient sensing pathways and decreased metabolic rate. The molecular mechanisms by which such restriction results in lengthened lifespan are as yet unclear (see[66] for some discussion); however, the behavior of many genes known to be involved in DNA repair is altered under conditions of caloric restriction. Several agents reported to have anti-aging properties have been shown to attenuate constitutive level of mTOR signaling, an evidence of reduction of metabolic activity, and concurrently to reduce constitutive level of DNA damage induced by endogenously generated reactive oxygen species.[67]
For example, increasing the gene dosage of the gene SIR-2, which regulates DNA packaging in the nematode worm Caenorhabditis elegans, can significantly extend lifespan.[68] The mammalian homolog of SIR-2 is known to induce downstream DNA repair factors involved in NHEJ, an activity that is especially promoted under conditions of caloric restriction.[69] Caloric restriction has been closely linked to the rate of base excision repair in the nuclear DNA of rodents,[70] although similar effects have not been observed in mitochondrial DNA.[71]
The C. elegans gene AGE-1, an upstream effector of DNA repair pathways, confers dramatically extended life span under free-feeding conditions but leads to a decrease in reproductive fitness under conditions of caloric restriction.[72] This observation supports the pleiotropy theory of the biological origins of aging, which suggests that genes conferring a large survival advantage early in life will be selected for even if they carry a corresponding disadvantage late in life.
Medicine and DNA repair modulation[edit]
Hereditary DNA repair disorders[edit]
Defects in the NER mechanism are responsible for several genetic disorders, including:
- Xeroderma pigmentosum: hypersensitivity to sunlight/UV, resulting in increased skin cancer incidence and premature aging
- Cockayne syndrome: hypersensitivity to UV and chemical agents
- Trichothiodystrophy: sensitive skin, brittle hair and nails
Mental retardation often accompanies the latter two disorders, suggesting increased vulnerability of developmental neurons.
Other DNA repair disorders include:
- Werner’s syndrome: premature aging and retarded growth
- Bloom’s syndrome: sunlight hypersensitivity, high incidence of malignancies (especially leukemias).
- Ataxia telangiectasia: sensitivity to ionizing radiation and some chemical agents
All of the above diseases are often called «segmental progerias» («accelerated aging diseases») because those affected appear elderly and experience aging-related diseases at an abnormally young age, while not manifesting all the symptoms of old age.
Other diseases associated with reduced DNA repair function include Fanconi anemia, hereditary breast cancer and hereditary colon cancer.
Cancer[edit]
Because of inherent limitations in the DNA repair mechanisms, if humans lived long enough, they would all eventually develop cancer.[73][74] There are at least 34 Inherited human DNA repair gene mutations that increase cancer risk. Many of these mutations cause DNA repair to be less effective than normal. In particular, Hereditary nonpolyposis colorectal cancer (HNPCC) is strongly associated with specific mutations in the DNA mismatch repair pathway. BRCA1 and BRCA2, two important genes whose mutations confer a hugely increased risk of breast cancer on carriers,[75] are both associated with a large number of DNA repair pathways, especially NHEJ and homologous recombination.
Cancer therapy procedures such as chemotherapy and radiotherapy work by overwhelming the capacity of the cell to repair DNA damage, resulting in cell death. Cells that are most rapidly dividing – most typically cancer cells – are preferentially affected. The side-effect is that other non-cancerous but rapidly dividing cells such as progenitor cells in the gut, skin, and hematopoietic system are also affected. Modern cancer treatments attempt to localize the DNA damage to cells and tissues only associated with cancer, either by physical means (concentrating the therapeutic agent in the region of the tumor) or by biochemical means (exploiting a feature unique to cancer cells in the body). In the context of therapies targeting DNA damage response genes, the latter approach has been termed ‘synthetic lethality’.[76]
Perhaps the most well-known of these ‘synthetic lethality’ drugs is the poly(ADP-ribose) polymerase 1 (PARP1) inhibitor olaparib, which was approved by the Food and Drug Administration in 2015 for the treatment in women of BRCA-defective ovarian cancer. Tumor cells with partial loss of DNA damage response (specifically, homologous recombination repair) are dependent on another mechanism – single-strand break repair – which is a mechanism consisting, in part, of the PARP1 gene product.[77] Olaparib is combined with chemotherapeutics to inhibit single-strand break repair induced by DNA damage caused by the co-administered chemotherapy. Tumor cells relying on this residual DNA repair mechanism are unable to repair the damage and hence are not able to survive and proliferate, whereas normal cells can repair the damage with the functioning homologous recombination mechanism.
Many other drugs for use against other residual DNA repair mechanisms commonly found in cancer are currently under investigation. However, synthetic lethality therapeutic approaches have been questioned due to emerging evidence of acquired resistance, achieved through rewiring of DNA damage response pathways and reversion of previously inhibited defects.[78]
DNA repair defects in cancer[edit]
It has become apparent over the past several years that the DNA damage response acts as a barrier to the malignant transformation of preneoplastic cells.[79] Previous studies have shown an elevated DNA damage response in cell-culture models with oncogene activation[80] and preneoplastic colon adenomas.[81] DNA damage response mechanisms trigger cell-cycle arrest, and attempt to repair DNA lesions or promote cell death/senescence if repair is not possible. Replication stress is observed in preneoplastic cells due to increased proliferation signals from oncogenic mutations. Replication stress is characterized by: increased replication initiation/origin firing; increased transcription and collisions of transcription-replication complexes; nucleotide deficiency; increase in reactive oxygen species (ROS).[82]
Replication stress, along with the selection for inactivating mutations in DNA damage response genes in the evolution of the tumor,[83] leads to downregulation and/or loss of some DNA damage response mechanisms, and hence loss of DNA repair and/or senescence/programmed cell death. In experimental mouse models, loss of DNA damage response-mediated cell senescence was observed after using a short hairpin RNA (shRNA) to inhibit the double-strand break response kinase ataxia telangiectasia (ATM), leading to increased tumor size and invasiveness.[81] Humans born with inherited defects in DNA repair mechanisms (for example, Li-Fraumeni syndrome) have a higher cancer risk.[84]
The prevalence of DNA damage response mutations differs across cancer types; for example, 30% of breast invasive carcinomas have mutations in genes involved in homologous recombination.[79] In cancer, downregulation is observed across all DNA damage response mechanisms (base excision repair (BER), nucleotide excision repair (NER), DNA mismatch repair (MMR), homologous recombination repair (HR), non-homologous end joining (NHEJ) and translesion DNA synthesis (TLS).[85] As well as mutations to DNA damage repair genes, mutations also arise in the genes responsible for arresting the cell cycle to allow sufficient time for DNA repair to occur, and some genes are involved in both DNA damage repair and cell cycle checkpoint control, for example ATM and checkpoint kinase 2 (CHEK2) – a tumor suppressor that is often absent or downregulated in non-small cell lung cancer.[86]
Genes involved in DNA damage response pathways and frequently mutated in cancer (HR = homologous recombination; NHEJ = non-homologous end joining; SSA = single-strand annealing; FA = fanconi anemia pathway; BER = base excision repair; NER = nucleotide excision repair; MMR = mismatch repair)
| HR | NHEJ | SSA | FA | BER | NER | MMR | |
|---|---|---|---|---|---|---|---|
| ATM | |||||||
| ATR | |||||||
| PAXIP | |||||||
| RPA | |||||||
| BRCA1 | |||||||
| BRCA2 | |||||||
| RAD51 | |||||||
| RFC | |||||||
| XRCC1 | |||||||
| PCNA | |||||||
| PARP1 | |||||||
| ERCC1 | |||||||
| MSH3 |
Epigenetic DNA repair defects in cancer[edit]
Classically, cancer has been viewed as a set of diseases that are driven by progressive genetic abnormalities that include mutations in tumour-suppressor genes and oncogenes, and chromosomal aberrations. However, it has become apparent that cancer is also driven by
epigenetic alterations.[87]
Epigenetic alterations refer to functionally relevant modifications to the genome that do not involve a change in the nucleotide sequence. Examples of such modifications are changes in DNA methylation (hypermethylation and hypomethylation) and histone modification,[88] changes in chromosomal architecture (caused by inappropriate expression of proteins such as HMGA2 or HMGA1)[89] and changes caused by microRNAs. Each of these epigenetic alterations serves to regulate gene expression without altering the underlying DNA sequence. These changes usually remain through cell divisions, last for multiple cell generations, and can be considered to be epimutations (equivalent to mutations).
While large numbers of epigenetic alterations are found in cancers, the epigenetic alterations in DNA repair genes, causing reduced expression of DNA repair proteins, appear to be particularly important. Such alterations are thought to occur early in progression to cancer and to be a likely cause of the genetic instability characteristic of cancers.[90][91][92]
Reduced expression of DNA repair genes causes deficient DNA repair. When DNA repair is deficient DNA damages remain in cells at a higher than usual level and these excess damages cause increased frequencies of mutation or epimutation. Mutation rates increase substantially in cells defective in DNA mismatch repair[93][94] or in homologous recombinational repair (HRR).[95] Chromosomal rearrangements and aneuploidy also increase in HRR defective cells.[96]
Higher levels of DNA damage not only cause increased mutation, but also cause increased epimutation. During repair of DNA double strand breaks, or repair of other DNA damages, incompletely cleared sites of repair can cause epigenetic gene silencing.[97][98]
Deficient expression of DNA repair proteins due to an inherited mutation can cause increased risk of cancer. Individuals with an inherited impairment in any of 34 DNA repair genes (see article DNA repair-deficiency disorder) have an increased risk of cancer, with some defects causing up to a 100% lifetime chance of cancer (e.g. p53 mutations).[99] However, such germline mutations (which cause highly penetrant cancer syndromes) are the cause of only about 1 percent of cancers.[100]
Frequencies of epimutations in DNA repair genes[edit]
A chart of common DNA damaging agents, examples of lesions they cause in DNA, and pathways used to repair these lesions. Also shown are many of the genes in these pathways, an indication of which genes are epigenetically regulated to have reduced (or increased) expression in various cancers. It also shows genes in the error-prone microhomology-mediated end joining pathway with increased expression in various cancers.
Deficiencies in DNA repair enzymes are occasionally caused by a newly arising somatic mutation in a DNA repair gene, but are much more frequently caused by epigenetic alterations that reduce or silence expression of DNA repair genes. For example, when 113 colorectal cancers were examined in sequence, only four had a missense mutation in the DNA repair gene MGMT, while the majority had reduced MGMT expression due to methylation of the MGMT promoter region (an epigenetic alteration).[101] Five different studies found that between 40% and 90% of colorectal cancers have reduced MGMT expression due to methylation of the MGMT promoter region.[102][103][104][105][106]
Similarly, out of 119 cases of mismatch repair-deficient colorectal cancers that lacked DNA repair gene PMS2 expression, PMS2 was deficient in 6 due to mutations in the PMS2 gene, while in 103 cases PMS2 expression was deficient because its pairing partner MLH1 was repressed due to promoter methylation (PMS2 protein is unstable in the absence of MLH1).[107] In the other 10 cases, loss of PMS2 expression was likely due to epigenetic overexpression of the microRNA, miR-155, which down-regulates MLH1.[108]
In a further example, epigenetic defects were found in various cancers (e.g. breast, ovarian, colorectal and head and neck). Two or three deficiencies in the expression of ERCC1, XPF or PMS2 occur simultaneously in the majority of 49 colon cancers evaluated by Facista et al.[109]
The chart in this section shows some frequent DNA damaging agents, examples of DNA lesions they cause, and the pathways that deal with these DNA damages. At least 169 enzymes are either directly employed in DNA repair or influence DNA repair processes.[110] Of these, 83 are directly employed in repairing the 5 types of DNA damages illustrated in the chart.
Some of the more well studied genes central to these repair processes are shown in the chart. The gene designations shown in red, gray or cyan indicate genes frequently epigenetically altered in various types of cancers. Wikipedia articles on each of the genes highlighted by red, gray or cyan describe the epigenetic alteration(s) and the cancer(s) in which these epimutations are found. Review articles,[111] and broad experimental survey articles[112][113] also document most of these epigenetic DNA repair deficiencies in cancers.
Red-highlighted genes are frequently reduced or silenced by epigenetic mechanisms in various cancers. When these genes have low or absent expression, DNA damages can accumulate. Replication errors past these damages (see translesion synthesis) can lead to increased mutations and, ultimately, cancer. Epigenetic repression of DNA repair genes in accurate DNA repair pathways appear to be central to carcinogenesis.
The two gray-highlighted genes RAD51 and BRCA2, are required for homologous recombinational repair. They are sometimes epigenetically over-expressed and sometimes under-expressed in certain cancers. As indicated in the Wikipedia articles on RAD51 and BRCA2, such cancers ordinarily have epigenetic deficiencies in other DNA repair genes. These repair deficiencies would likely cause increased unrepaired DNA damages. The over-expression of RAD51 and BRCA2 seen in these cancers may reflect selective pressures for compensatory RAD51 or BRCA2 over-expression and increased homologous recombinational repair to at least partially deal with such excess DNA damages. In those cases where RAD51 or BRCA2 are under-expressed, this would itself lead to increased unrepaired DNA damages. Replication errors past these damages (see translesion synthesis) could cause increased mutations and cancer, so that under-expression of RAD51 or BRCA2 would be carcinogenic in itself.
Cyan-highlighted genes are in the microhomology-mediated end joining (MMEJ) pathway and are up-regulated in cancer. MMEJ is an additional error-prone inaccurate repair pathway for double-strand breaks. In MMEJ repair of a double-strand break, an homology of 5–25 complementary base pairs between both paired strands is sufficient to align the strands, but mismatched ends (flaps) are usually present. MMEJ removes the extra nucleotides (flaps) where strands are joined, and then ligates the strands to create an intact DNA double helix. MMEJ almost always involves at least a small deletion, so that it is a mutagenic pathway.[24] FEN1, the flap endonuclease in MMEJ, is epigenetically increased by promoter hypomethylation and is over-expressed in the majority of cancers of the breast,[114] prostate,[115] stomach,[116][117] neuroblastomas,[118] pancreas,[119] and lung.[120] PARP1 is also over-expressed when its promoter region ETS site is epigenetically hypomethylated, and this contributes to progression to endometrial cancer[121] and BRCA-mutated serous ovarian cancer.[122] Other genes in the MMEJ pathway are also over-expressed in a number of cancers (see MMEJ for summary), and are also shown in cyan.
Genome-wide distribution of DNA repair in human somatic cells[edit]
Differential activity of DNA repair pathways across various regions of the human genome causes mutations to be very unevenly distributed within tumor genomes.[123][124] In particular, the gene-rich, early-replicating regions of the human genome exhibit lower mutation frequencies than the gene-poor, late-replicating heterochromatin. One mechanism underlying this involves the histone modification H3K36me3, which can recruit mismatch repair proteins,[125] thereby lowering mutation rates in H3K36me3-marked regions.[126] Another important mechanism concerns nucleotide excision repair, which can be recruited by the transcription machinery, lowering somatic mutation rates in active genes[124] and other open chromatin regions.[127]
Epigenetic alterations due to DNA repair[edit]
Damage to DNA is very common and is constantly being repaired. Epigenetic alterations can accompany DNA repair of oxidative damage or double-strand breaks. In human cells, oxidative DNA damage occurs about 10,000 times a day and DNA double-strand breaks occur about 10 to 50 times a cell cycle in somatic replicating cells (see DNA damage (naturally occurring)). The selective advantage of DNA repair is to allow the cell to survive in the face of DNA damage. The selective advantage of epigenetic alterations that occur with DNA repair is not clear.
Repair of oxidative DNA damage can alter epigenetic markers[edit]
In the steady state (with endogenous damages occurring and being repaired), there are about 2,400 oxidatively damaged guanines that form 8-oxo-2′-deoxyguanosine (8-OHdG) in the average mamalian cell DNA.[128] 8-OHdG constitutes about 5% of the oxidative damages commonly present in DNA.[129] The oxidized guanines do not occur randomly among all guanines in DNA. There is a sequence preference for the guanine at a methylated CpG site (a cytosine followed by guanine along its 5′ → 3′ direction and where the cytosine is methylated (5-mCpG)).[130] A 5-mCpG site has the lowest ionization potential for guanine oxidation.
Initiation of DNA demethylation at a CpG site. In adult somatic cells DNA methylation typically occurs in the context of CpG dinucleotides (CpG sites), forming 5-methylcytosine-pG, or 5mCpG. Reactive oxygen species (ROS) may attack guanine at the dinucleotide site, forming 8-hydroxy-2′-deoxyguanosine (8-OHdG), and resulting in a 5mCp-8-OHdG dinucleotide site. The base excision repair enzyme OGG1 targets 8-OHdG and binds to the lesion without immediate excision. OGG1, present at a 5mCp-8-OHdG site recruits TET1 and TET1 oxidizes the 5mC adjacent to the 8-OHdG. This initiates demethylation of 5mC.[131]
Oxidized guanine has mispairing potential and is mutagenic.[132] Oxoguanine glycosylase (OGG1) is the primary enzyme responsible for the excision of the oxidized guanine during DNA repair. OGG1 finds and binds to an 8-OHdG within a few seconds.[133] However, OGG1 does not immediately excise 8-OHdG. In HeLa cells half maximum removal of 8-OHdG occurs in 30 minutes,[134] and in irradiated mice, the 8-OHdGs induced in the mouse liver are removed with a half-life of 11 minutes.[129]
When OGG1 is present at an oxidized guanine within a methylated CpG site it recruits TET1 to the 8-OHdG lesion (see Figure). This allows TET1 to demethylate an adjacent methylated cytosine. Demethylation of cytosine is an epigenetic alteration.
As an example, when human mammary epithelial cells were treated with H2O2 for six hours, 8-OHdG increased about 3.5-fold in DNA and this caused about 80% demethylation of the 5-methylcytosines in the genome.[131] Demethylation of CpGs in a gene promoter by TET enzyme activity increases transcription of the gene into messenger RNA.[135] In cells treated with H2O2, one particular gene was examined, BACE1.[131] The methylation level of the BACE1 CpG island was reduced (an epigenetic alteration) and this allowed about 6.5 fold increase of expression of BACE1 messenger RNA.
While six-hour incubation with H2O2 causes considerable demethylation of 5-mCpG sites, shorter times of H2O2 incubation appear to promote other epigenetic alterations. Treatment of cells with H2O2 for 30 minutes causes the mismatch repair protein heterodimer MSH2-MSH6 to recruit DNA methyltransferase 1 (DNMT1) to sites of some kinds of oxidative DNA damage.[136] This could cause increased methylation of cytosines (epigenetic alterations) at these locations.
Jiang et al.[137] treated HEK 293 cells with agents causing oxidative DNA damage, (potassium bromate (KBrO3) or potassium chromate (K2CrO4)). Base excision repair (BER) of oxidative damage occurred with the DNA repair enzyme polymerase beta localizing to oxidized guanines. Polymerase beta is the main human polymerase in short-patch BER of oxidative DNA damage. Jiang et al.[137] also found that polymerase beta recruited the DNA methyltransferase protein DNMT3b to BER repair sites. They then evaluated the methylation pattern at the single nucleotide level in a small region of DNA including the promoter region and the early transcription region of the BRCA1 gene. Oxidative DNA damage from bromate modulated the DNA methylation pattern (caused epigenetic alterations) at CpG sites within the region of DNA studied. In untreated cells, CpGs located at −189, −134, −29, −19, +16, and +19 of the BRCA1 gene had methylated cytosines (where numbering is from the messenger RNA transcription start site, and negative numbers indicate nucleotides in the upstream promoter region). Bromate treatment-induced oxidation resulted in the loss of cytosine methylation at −189, −134, +16 and +19 while also leading to the formation of new methylation at the CpGs located at −80, −55, −21 and +8 after DNA repair was allowed.
Homologous recombinational repair alters epigenetic markers[edit]
At least four articles report the recruitment of DNA methyltransferase 1 (DNMT1) to sites of DNA double-strand breaks.[138][139][140][141] During homologous recombinational repair (HR) of the double-strand break, the involvement of DNMT1 causes the two repaired strands of DNA to have different levels of methylated cytosines. One strand becomes frequently methylated at about 21 CpG sites downstream of the repaired double-strand break. The other DNA strand loses methylation at about six CpG sites that were previously methylated downstream of the double-strand break, as well as losing methylation at about five CpG sites that were previously methylated upstream of the double-strand break. When the chromosome is replicated, this gives rise to one daughter chromosome that is heavily methylated downstream of the previous break site and one that is unmethylated in the region both upstream and downstream of the previous break site. With respect to the gene that was broken by the double-strand break, half of the progeny cells express that gene at a high level and in the other half of the progeny cells expression of that gene is repressed. When clones of these cells were maintained for three years, the new methylation patterns were maintained over that time period.[142]
In mice with a CRISPR-mediated homology-directed recombination insertion in their genome there were a large number of increased methylations of CpG sites within the double-strand break-associated insertion.[143]
Non-homologous end joining can cause some epigenetic marker alterations[edit]
Non-homologous end joining (NHEJ) repair of a double-strand break can cause a small number of demethylations of pre-existing cytosine DNA methylations downstream of the repaired double-strand break.[139] Further work by Allen et al.[144] showed that NHEJ of a DNA double-strand break in a cell could give rise to some progeny cells having repressed expression of the gene harboring the initial double-strand break and some progeny having high expression of that gene due to epigenetic alterations associated with NHEJ repair. The frequency of epigenetic alterations causing repression of a gene after an NHEJ repair of a DNA double-strand break in that gene may be about 0.9%.[140]
Evolution[edit]
The basic processes of DNA repair are highly conserved among both prokaryotes and eukaryotes and even among bacteriophages (viruses which infect bacteria); however, more complex organisms with more complex genomes have correspondingly more complex repair mechanisms.[145] The ability of a large number of protein structural motifs to catalyze relevant chemical reactions has played a significant role in the elaboration of repair mechanisms during evolution. For an extremely detailed review of hypotheses relating to the evolution of DNA repair, see.[146]
The fossil record indicates that single-cell life began to proliferate on the planet at some point during the Precambrian period, although exactly when recognizably modern life first emerged is unclear. Nucleic acids became the sole and universal means of encoding genetic information, requiring DNA repair mechanisms that in their basic form have been inherited by all extant life forms from their common ancestor. The emergence of Earth’s oxygen-rich atmosphere (known as the «oxygen catastrophe») due to photosynthetic organisms, as well as the presence of potentially damaging free radicals in the cell due to oxidative phosphorylation, necessitated the evolution of DNA repair mechanisms that act specifically to counter the types of damage induced by oxidative stress.
Rate of evolutionary change[edit]
On some occasions, DNA damage is not repaired or is repaired by an error-prone mechanism that results in a change from the original sequence. When this occurs, mutations may propagate into the genomes of the cell’s progeny. Should such an event occur in a germ line cell that will eventually produce a gamete, the mutation has the potential to be passed on to the organism’s offspring. The rate of evolution in a particular species (or, in a particular gene) is a function of the rate of mutation. As a consequence, the rate and accuracy of DNA repair mechanisms have an influence over the process of evolutionary change.[147] DNA damage protection and repair does not influence the rate of adaptation by gene regulation and by recombination and selection of alleles. On the other hand, DNA damage repair and protection does influence the rate of accumulation of irreparable, advantageous, code expanding, inheritable mutations, and slows down the evolutionary mechanism for expansion of the genome of organisms with new functionalities. The tension between evolvability and mutation repair and protection needs further investigation.
Technology[edit]
A technology named clustered regularly interspaced short palindromic repeat (shortened to CRISPR-Cas9) was discovered in 2012. The new technology allows anyone with molecular biology training to alter the genes of any species with precision, by inducing DNA damage at a specific point and then altering DNA repair mechanisms to insert new genes.[148] It is cheaper, more efficient, and more precise than other technologies. With the help of CRISPR–Cas9, parts of a genome can be edited by scientists by removing, adding, or altering parts in a DNA sequence.
See also[edit]
- Accelerated aging disease
- Aging DNA
- Cell cycle
- DNA damage (naturally occurring)
- DNA damage theory of aging
- DNA replication
- Direct DNA damage
- Gene therapy
- Human mitochondrial genetics
- Indirect DNA damage
- Life extension
- Progeria
- REPAIRtoire
- Senescence
- SiDNA
- The scientific journal DNA Repair under Mutation Research
References[edit]
- ^ «Nature Reviews Series: DNA damage». Nature Reviews Molecular Cell Biology. 5 July 2017. Retrieved 7 November 2018.
- ^ a b Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J (2004). Molecular Biology of the Cell (5th ed.). New York: WH Freeman. p. 963.
- ^ a b Acharya PV (1971). «The isolation and partial characterization of age-correlated oligo-deoxyribo-ribonucleotides with covalently linked aspartyl-glutamyl polypeptides». Johns Hopkins Medical Journal. Supplement (1): 254–60. PMID 5055816.
- ^ a b Bjorksten J, Acharya PV, Ashman S, Wetlaufer DB (July 1971). «Gerogenic fractions in the tritiated rat». Journal of the American Geriatrics Society. 19 (7): 561–74. doi:10.1111/j.1532-5415.1971.tb02577.x. PMID 5106728. S2CID 33154242.
- ^ Browner WS, Kahn AJ, Ziv E, Reiner AP, Oshima J, Cawthon RM, et al. (December 2004). «The genetics of human longevity». The American Journal of Medicine. 117 (11): 851–60. CiteSeerX 10.1.1.556.6874. doi:10.1016/j.amjmed.2004.06.033. PMID 15589490.
- ^ Broad WJ (7 October 2015). «Nobel Prize in Chemistry Awarded to Tomas Lindahl, Paul Modrich and Aziz Sancar for DNA Studies». The New York Times. Retrieved 7 October 2015.
- ^ Staff (7 October 2015). «The Nobel Prize in Chemistry 2015 – DNA repair – providing chemical stability for life» (PDF). Nobel Prize. Retrieved 7 October 2015.
- ^ Roulston A, Marcellus RC, Branton PE (1999). «Viruses and apoptosis». Annual Review of Microbiology. 53: 577–628. doi:10.1146/annurev.micro.53.1.577. PMID 10547702.
- ^ Madigan MT, Martino JM (2006). Brock Biology of Microorganisms (11th ed.). Pearson. p. 136. ISBN 978-0-13-196893-6.
- ^ Ohta T, Tokishita SI, Mochizuki K, Kawase J, Sakahira M, Yamagata H (2006). «UV Sensitivity and Mutagenesis of the Extremely Thermophilic Eubacterium Thermus thermophilus HB27». Genes and Environment. 28 (2): 56–61. doi:10.3123/jemsge.28.56.
- ^ Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z (September 2006). «Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants». Cell Cycle. 5 (17): 1940–45. doi:10.4161/cc.5.17.3191. PMC 3488278. PMID 16940754.
- ^ Braig M, Schmitt CA (March 2006). «Oncogene-induced senescence: putting the brakes on tumor development». Cancer Research. 66 (6): 2881–84. doi:10.1158/0008-5472.CAN-05-4006. PMID 16540631.
- ^ Lynch MD (February 2006). «How does cellular senescence prevent cancer?». DNA and Cell Biology. 25 (2): 69–78. doi:10.1089/dna.2006.25.69. PMID 16460230.
- ^ Campisi J, d’Adda di Fagagna F (September 2007). «Cellular senescence: when bad things happen to good cells». Nature Reviews. Molecular Cell Biology. 8 (9): 729–40. doi:10.1038/nrm2233. PMID 17667954. S2CID 15664931.
- ^ a b c Best BP (June 2009). «Nuclear DNA damage as a direct cause of aging» (PDF). Rejuvenation Research. 12 (3): 199–208. CiteSeerX 10.1.1.318.738. doi:10.1089/rej.2009.0847. PMID 19594328. Archived from the original (PDF) on 15 November 2017. Retrieved 29 September 2009.
- ^ Sancar A (June 2003). «Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors». Chemical Reviews. 103 (6): 2203–37. doi:10.1021/cr0204348. PMID 12797829.
- ^ Lucas-Lledó JI, Lynch M (May 2009). «Evolution of mutation rates: phylogenomic analysis of the photolyase/cryptochrome family». Molecular Biology and Evolution. 26 (5): 1143–53. doi:10.1093/molbev/msp029. PMC 2668831. PMID 19228922.
- ^ a b c Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R (2004). Molecular Biology of the Gene (5th ed.). Pearson Benjamin Cummings; CSHL Press. Ch. 9, 10. OCLC 936762772.
- ^ Volkert MR (1988). «Adaptive response of Escherichia coli to alkylation damage». Environmental and Molecular Mutagenesis. 11 (2): 241–55. doi:10.1002/em.2850110210. PMID 3278898. S2CID 24722637.
- ^ a b c Willey J, Sherwood L, Woolverton C (2014). Prescott’s Microbiology. New York: McGraw Hill. p. 381. ISBN 978-0-07-3402-40-6.
- ^ Russell P (2018). i Genetics. Chennai: Pearson. p. 186. ISBN 978-93-325-7162-4.
- ^ a b c d Reardon JT, Sancar A (2006). «Purification and characterization of Escherichia coli and human nucleotide excision repair enzyme systems». Methods in Enzymology. 408: 189–213. doi:10.1016/S0076-6879(06)08012-8. ISBN 9780121828134. PMID 16793370.
- ^ Berg M, Tymoczko J, Stryer L (2012). Biochemistry 7th edition. New York: W.H. Freeman and Company. p. 840. ISBN 9781429229364.
- ^ a b Liang L, Deng L, Chen Y, Li GC, Shao C, Tischfield JA (September 2005). «Modulation of DNA end joining by nuclear proteins». The Journal of Biological Chemistry. 280 (36): 31442–49. doi:10.1074/jbc.M503776200. PMID 16012167.
- ^ Wilson TE, Grawunder U, Lieber MR (July 1997). «Yeast DNA ligase IV mediates non-homologous DNA end joining». Nature. 388 (6641): 495–98. Bibcode:1997Natur.388..495W. doi:10.1038/41365. PMID 9242411. S2CID 4422938.
- ^ Moore JK, Haber JE (May 1996). «Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae». Molecular and Cellular Biology. 16 (5): 2164–73. doi:10.1128/mcb.16.5.2164. PMC 231204. PMID 8628283.
- ^ Boulton SJ, Jackson SP (September 1996). «Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways». The EMBO Journal. 15 (18): 5093–103. doi:10.1002/j.1460-2075.1996.tb00890.x. PMC 452249. PMID 8890183.
- ^ Wilson TE, Lieber MR (August 1999). «Efficient processing of DNA ends during yeast nonhomologous end joining. Evidence for a DNA polymerase beta (Pol4)-dependent pathway». The Journal of Biological Chemistry. 274 (33): 23599–609. doi:10.1074/jbc.274.33.23599. PMID 10438542.
- ^ Budman J, Chu G (February 2005). «Processing of DNA for nonhomologous end-joining by cell-free extract». The EMBO Journal. 24 (4): 849–60. doi:10.1038/sj.emboj.7600563. PMC 549622. PMID 15692565.
- ^ Wang H, Perrault AR, Takeda Y, Qin W, Wang H, Iliakis G (September 2003). «Biochemical evidence for Ku-independent backup pathways of NHEJ». Nucleic Acids Research. 31 (18): 5377–88. doi:10.1093/nar/gkg728. PMC 203313. PMID 12954774.
- ^ Jung D, Alt FW (January 2004). «Unraveling V(D)J recombination; insights into gene regulation». Cell. 116 (2): 299–311. doi:10.1016/S0092-8674(04)00039-X. PMID 14744439. S2CID 16890458.
- ^ a b Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, et al. (May 2013). «Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells». Proceedings of the National Academy of Sciences of the United States of America. 110 (19): 7720–25. Bibcode:2013PNAS..110.7720T. doi:10.1073/pnas.1213431110. PMC 3651503. PMID 23610439.
- ^ Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC (March 2015). «Homology and enzymatic requirements of microhomology-dependent alternative end joining». Cell Death & Disease. 6 (3): e1697. doi:10.1038/cddis.2015.58. PMC 4385936. PMID 25789972.
- ^ Decottignies A (2013). «Alternative end-joining mechanisms: a historical perspective». Frontiers in Genetics. 4: 48. doi:10.3389/fgene.2013.00048. PMC 3613618. PMID 23565119.
- ^ Zahradka K, Slade D, Bailone A, Sommer S, Averbeck D, Petranovic M, et al. (October 2006). «Reassembly of shattered chromosomes in Deinococcus radiodurans». Nature. 443 (7111): 569–73. Bibcode:2006Natur.443..569Z. doi:10.1038/nature05160. PMID 17006450. S2CID 4412830.
- ^ Stenerlöw B, Karlsson KH, Cooper B, Rydberg B. «Measurement of prompt DNA double-strand breaks in mammalian cells without including heat-labile sites: results for cells deficient in nonhomologous end joining». Radiat Res. 2003 Apr;159(4):502–10. doi:10.1667/0033-7587(2003)159[0502:mopdds2.0.co;2] PMID 12643795.
- ^ Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC (March 2009). «Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance». Microbiology and Molecular Biology Reviews. 73 (1): 134–54. doi:10.1128/MMBR.00034-08. PMC 2650891. PMID 19258535.
- ^ Colis LC, Raychaudhury P, Basu AK (August 2008). «Mutational specificity of gamma-radiation-induced guanine-thymine and thymine-guanine intrastrand cross-links in mammalian cells and translesion synthesis past the guanine-thymine lesion by human DNA polymerase eta». Biochemistry. 47 (31): 8070–79. doi:10.1021/bi800529f. PMC 2646719. PMID 18616294.
- ^ Raychaudhury P, Basu AK (March 2011). «Genetic requirement for mutagenesis of the G[8,5-Me]T cross-link in Escherichia coli: DNA polymerases IV and V compete for error-prone bypass». Biochemistry. 50 (12): 2330–38. doi:10.1021/bi102064z. PMC 3062377. PMID 21302943.
- ^ «Translesion Synthesis». Research.chem.psu.edu. Archived from the original on 10 March 2012. Retrieved 14 August 2012.
- ^ Wang Z (July 2001). «Translesion synthesis by the UmuC family of DNA polymerases». Mutation Research. 486 (2): 59–70. doi:10.1016/S0921-8777(01)00089-1. PMID 11425512.
- ^ a b c Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. (2006). DNA Repair and Mutagenesis, part 3. ASM Press. 2nd ed.
- ^ Liu B, Yip RK, Zhou Z (November 2012). «Chromatin remodeling, DNA damage repair and aging». Current Genomics. 13 (7): 533–47. doi:10.2174/138920212803251373. PMC 3468886. PMID 23633913.
- ^ Halicka HD, Zhao H, Podhorecka M, Traganos F, Darzynkiewicz Z (July 2009). «Cytometric detection of chromatin relaxation, an early reporter of DNA damage response». Cell Cycle. 8 (14): 2233–37. doi:10.4161/cc.8.14.8984. PMC 3856216. PMID 19502789.
- ^ a b c Sellou H, Lebeaupin T, Chapuis C, Smith R, Hegele A, Singh HR, et al. (December 2016). «The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage». Molecular Biology of the Cell. 27 (24): 3791–99. doi:10.1091/mbc.E16-05-0269. PMC 5170603. PMID 27733626.
- ^ a b Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, et al. (September 2016). «JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks». Cell Reports. 16 (10): 2641–50. doi:10.1016/j.celrep.2016.08.006. PMC 5089070. PMID 27568560.
- ^ a b Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG (January 2008). «PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites». The Journal of Biological Chemistry. 283 (2): 1197–208. doi:10.1074/jbc.M706734200. PMID 18025084.
- ^ a b c Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (March 1998). «DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139». The Journal of Biological Chemistry. 273 (10): 5858–68. doi:10.1074/jbc.273.10.5858. PMID 9488723.
- ^ Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J (November 2007). «RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins». Cell. 131 (5): 887–900. doi:10.1016/j.cell.2007.09.040. PMID 18001824. S2CID 14232192.
- ^ Luijsterburg MS, Acs K, Ackermann L, Wiegant WW, Bekker-Jensen S, Larsen DH, et al. (May 2012). «A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure». The EMBO Journal. 31 (11): 2511–27. doi:10.1038/emboj.2012.104. PMC 3365417. PMID 22531782.
- ^ a b Luijsterburg MS, Goedhart J, Moser J, Kool H, Geverts B, Houtsmuller AB, et al. (August 2007). «Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC». Journal of Cell Science. 120 (Pt 15): 2706–16. doi:10.1242/jcs.008367. PMID 17635991.
- ^ a b Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, et al. (October 2012). «PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1». The Journal of Cell Biology. 199 (2): 235–49. doi:10.1083/jcb.201112132. PMC 3471223. PMID 23045548.
- ^ Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP (January 2006). «ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks». Nature Cell Biology. 8 (1): 37–45. doi:10.1038/ncb1337. PMID 16327781. S2CID 9797133.
- ^ Bakkenist CJ, Kastan MB (January 2003). «DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation». Nature. 421 (6922): 499–506. Bibcode:2003Natur.421..499B. doi:10.1038/nature01368. PMID 12556884. S2CID 4403303.
- ^ Wei Q, Li L, Chen D (2007). DNA Repair, Genetic Instability, and Cancer. World Scientific. ISBN 978-981-270-014-8.[page needed]
- ^ Schonthal AH (2004). Checkpoint Controls and Cancer. Humana Press. ISBN 978-1-58829-500-2.[page needed]
- ^ Gartel AL, Tyner AL (June 2002). «The role of the cyclin-dependent kinase inhibitor p21 in apoptosis». Molecular Cancer Therapeutics. 1 (8): 639–49. PMID 12479224.
- ^ Janion C (2001). «Some aspects of the SOS response system—a critical survey». Acta Biochimica Polonica. 48 (3): 599–610. doi:10.18388/abp.2001_3894. PMID 11833768.
- ^ a b Erill I, Campoy S, Barbé J (November 2007). «Aeons of distress: an evolutionary perspective on the bacterial SOS response». FEMS Microbiology Reviews. 31 (6): 637–56. doi:10.1111/j.1574-6976.2007.00082.x. PMID 17883408.
- ^ Schlacher K, Pham P, Cox MM, Goodman MF (February 2006). «Roles of DNA polymerase V and RecA protein in SOS damage-induced mutation». Chemical Reviews. 106 (2): 406–19. doi:10.1021/cr0404951. PMID 16464012.
- ^ Fry RC, Begley TJ, Samson LD (2004). «Genome-wide responses to DNA-damaging agents». Annual Review of Microbiology. 59: 357–77. doi:10.1146/annurev.micro.59.031805.133658. PMID 16153173.
- ^ Espejel S, Martín M, Klatt P, Martín-Caballero J, Flores JM, Blasco MA (May 2004). «Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice». EMBO Reports. 5 (5): 503–09. doi:10.1038/sj.embor.7400127. PMC 1299048. PMID 15105825.
- ^ de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, et al. (May 2002). «Premature aging in mice deficient in DNA repair and transcription». Science. 296 (5571): 1276–79. Bibcode:2002Sci…296.1276D. doi:10.1126/science.1070174. PMID 11950998. S2CID 41930529.
- ^ Dollé ME, Busuttil RA, Garcia AM, Wijnhoven S, van Drunen E, Niedernhofer LJ, et al. (April 2006). «Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice». Mutation Research. 596 (1–2): 22–35. doi:10.1016/j.mrfmmm.2005.11.008. PMID 16472827.
- ^ Kobayashi Y, Narumi I, Satoh K, Funayama T, Kikuchi M, Kitayama S, Watanabe H (November 2004). «Radiation response mechanisms of the extremely radioresistant bacterium Deinococcus radiodurans». Uchu Seibutsu Kagaku. 18 (3): 134–35. PMID 15858357.
- ^ Spindler SR (September 2005). «Rapid and reversible induction of the longevity, anticancer and genomic effects of caloric restriction». Mechanisms of Ageing and Development. 126 (9): 960–66. doi:10.1016/j.mad.2005.03.016. PMID 15927235. S2CID 7067036.
- ^ Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, Wu JM, Darzynkiewicz Z (December 2012). «Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage- signaling». Aging. 4 (12): 952–65. doi:10.18632/aging.100521. PMC 3615161. PMID 23363784.
- ^ Tissenbaum HA, Guarente L (March 2001). «Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans». Nature. 410 (6825): 227–30. Bibcode:2001Natur.410..227T. doi:10.1038/35065638. PMID 11242085. S2CID 4356885.
- ^ Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. (July 2004). «Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase». Science. 305 (5682): 390–92. Bibcode:2004Sci…305..390C. doi:10.1126/science.1099196. PMID 15205477. S2CID 33503081.
- ^ Cabelof DC, Yanamadala S, Raffoul JJ, Guo Z, Soofi A, Heydari AR (March 2003). «Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline». DNA Repair. 2 (3): 295–307. doi:10.1016/S1568-7864(02)00219-7. PMID 12547392.
- ^ Stuart JA, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA (March 2004). «Mitochondrial and nuclear DNA base excision repair are affected differently by caloric restriction». FASEB Journal. 18 (3): 595–97. doi:10.1096/fj.03-0890fje. PMID 14734635. S2CID 43118901.
- ^ Walker DW, McColl G, Jenkins NL, Harris J, Lithgow GJ (May 2000). «Evolution of lifespan in C. elegans». Nature. 405 (6784): 296–97. doi:10.1038/35012693. PMID 10830948. S2CID 4402039.
- ^ Johnson G (28 December 2010). «Unearthing Prehistoric Tumors, and Debate». The New York Times.
If we lived long enough, sooner or later we all would get cancer.
- ^ Alberts B, Johnson A, Lewis J, et al. (2002). «The Preventable Causes of Cancer». Molecular biology of the cell (4th ed.). New York: Garland Science. ISBN 978-0-8153-4072-0.
A certain irreducible background incidence of cancer is to be expected regardless of circumstances: mutations can never be absolutely avoided, because they are an inescapable consequence of fundamental limitations on the accuracy of DNA replication, as discussed in Chapter 5. If a human could live long enough, it is inevitable that at least one of his or her cells would eventually accumulate a set of mutations sufficient for cancer to develop.
- ^ Friedenson B (August 2007). «The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers». BMC Cancer. 7: 152. doi:10.1186/1471-2407-7-152. PMC 1959234. PMID 17683622.
- ^ Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, Turchi JJ (April 2016). «DNA repair targeted therapy: The past or future of cancer treatment?». Pharmacology & Therapeutics. 160: 65–83. doi:10.1016/j.pharmthera.2016.02.003. PMC 4811676. PMID 26896565.
- ^ Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. (April 2005). «Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase». Nature. 434 (7035): 913–17. Bibcode:2005Natur.434..913B. doi:10.1038/nature03443. PMID 15829966. S2CID 4391043.
- ^ Goldstein M, Kastan MB (2015). «The DNA damage response: implications for tumor responses to radiation and chemotherapy». Annual Review of Medicine. 66: 129–43. doi:10.1146/annurev-med-081313-121208. PMID 25423595.
- ^ a b Jeggo PA, Pearl LH, Carr AM (January 2016). «DNA repair, genome stability and cancer: a historical perspective» (PDF). Nature Reviews. Cancer. 16 (1): 35–42. doi:10.1038/nrc.2015.4. PMID 26667849. S2CID 14941857.
- ^ Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, et al. (April 2005). «DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis». Nature. 434 (7035): 864–70. Bibcode:2005Natur.434..864B. doi:10.1038/nature03482. PMID 15829956. S2CID 4398393.
- ^ a b Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. (November 2006). «Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints». Nature. 444 (7119): 633–37. Bibcode:2006Natur.444..633B. doi:10.1038/nature05268. PMID 17136093. S2CID 4406956.
- ^ Gaillard H, García-Muse T, Aguilera A (May 2015). «Replication stress and cancer». Nature Reviews. Cancer. 15 (5): 276–89. doi:10.1038/nrc3916. hdl:10261/123721. PMID 25907220. S2CID 11342123.
- ^ Halazonetis TD, Gorgoulis VG, Bartek J (March 2008). «An oncogene-induced DNA damage model for cancer development». Science. 319 (5868): 1352–55. Bibcode:2008Sci…319.1352H. doi:10.1126/science.1140735. PMID 18323444. S2CID 16426080.
- ^ de Boer J, Hoeijmakers JH (March 2000). «Nucleotide excision repair and human syndromes» (PDF). Carcinogenesis. 21 (3): 453–60. doi:10.1093/carcin/21.3.453. PMID 10688865.
- ^ Broustas CG, Lieberman HB (February 2014). «DNA damage response genes and the development of cancer metastasis». Radiation Research. 181 (2): 111–30. Bibcode:2014RadR..181..111B. doi:10.1667/RR13515.1. PMC 4064942. PMID 24397478.
- ^ Zhang P, Wang J, Gao W, Yuan BZ, Rogers J, Reed E (May 2004). «CHK2 kinase expression is down-regulated due to promoter methylation in non-small cell lung cancer». Molecular Cancer. 3 (4): 14. doi:10.1186/1476-4598-3-14. PMC 419366. PMID 15125777.
- ^ Baylin SB, Ohm JE (February 2006). «Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction?». Nature Reviews. Cancer. 6 (2): 107–16. doi:10.1038/nrc1799. PMID 16491070. S2CID 2514545.
- ^ Kanwal R, Gupta S (April 2012). «Epigenetic modifications in cancer». Clinical Genetics. 81 (4): 303–11. doi:10.1111/j.1399-0004.2011.01809.x. PMC 3590802. PMID 22082348.
- ^ Baldassarre G, Battista S, Belletti B, Thakur S, Pentimalli F, Trapasso F, et al. (April 2003). «Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma». Molecular and Cellular Biology. 23 (7): 2225–38. doi:10.1128/MCB.23.7.2225-2238.2003. PMC 150734. PMID 12640109.
- ^ Jacinto FV, Esteller M (July 2007). «Mutator pathways unleashed by epigenetic silencing in human cancer». Mutagenesis. 22 (4): 247–53. doi:10.1093/mutage/gem009. PMID 17412712.
- ^ Lahtz C, Pfeifer GP (February 2011). «Epigenetic changes of DNA repair genes in cancer». Journal of Molecular Cell Biology. 3 (1): 51–58. doi:10.1093/jmcb/mjq053. PMC 3030973. PMID 21278452. Epigenetic changes of DNA repair genes in cancer
- ^ Bernstein C, Nfonsam V, Prasad AR, Bernstein H (March 2013). «Epigenetic field defects in progression to cancer». World Journal of Gastrointestinal Oncology. 5 (3): 43–49. doi:10.4251/wjgo.v5.i3.43. PMC 3648662. PMID 23671730.
- ^ Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (April 1997). «Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2». Proceedings of the National Academy of Sciences of the United States of America. 94 (7): 3122–27. Bibcode:1997PNAS…94.3122N. doi:10.1073/pnas.94.7.3122. PMC 20332. PMID 9096356.
- ^ Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM (December 2006). «Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6». Carcinogenesis. 27 (12): 2402–08. doi:10.1093/carcin/bgl079. PMC 2612936. PMID 16728433.
- ^ Tutt AN, van Oostrom CT, Ross GM, van Steeg H, Ashworth A (March 2002). «Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation». EMBO Reports. 3 (3): 255–60. doi:10.1093/embo-reports/kvf037. PMC 1084010. PMID 11850397.
- ^ German J (March 1969). «Bloom’s syndrome. I. Genetical and clinical observations in the first twenty-seven patients». American Journal of Human Genetics. 21 (2): 196–227. PMC 1706430. PMID 5770175.
- ^ O’Hagan HM, Mohammad HP, Baylin SB (August 2008). «Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island». PLOS Genetics. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- ^ Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, et al. (July 2007). «DNA damage, homology-directed repair, and DNA methylation». PLOS Genetics. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- ^ Malkin D (April 2011). «Li-fraumeni syndrome». Genes & Cancer. 2 (4): 475–84. doi:10.1177/1947601911413466. PMC 3135649. PMID 21779515.
- ^ Fearon ER (November 1997). «Human cancer syndromes: clues to the origin and nature of cancer». Science. 278 (5340): 1043–50. Bibcode:1997Sci…278.1043F. doi:10.1126/science.278.5340.1043. PMID 9353177.
- ^ Halford S, Rowan A, Sawyer E, Talbot I, Tomlinson I (June 2005). «O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G:C>A:T transitions». Gut. 54 (6): 797–802. doi:10.1136/gut.2004.059535. PMC 1774551. PMID 15888787.
- ^ Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, et al. (September 2005). «MGMT promoter methylation and field defect in sporadic colorectal cancer». Journal of the National Cancer Institute. 97 (18): 1330–38. doi:10.1093/jnci/dji275. PMID 16174854.
- ^ Psofaki V, Kalogera C, Tzambouras N, Stephanou D, Tsianos E, Seferiadis K, Kolios G (July 2010). «Promoter methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal adenomas». World Journal of Gastroenterology. 16 (28): 3553–60. doi:10.3748/wjg.v16.i28.3553. PMC 2909555. PMID 20653064.
- ^ Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Park CS, et al. (October 2011). «Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence». Langenbeck’s Archives of Surgery. 396 (7): 1017–26. doi:10.1007/s00423-011-0812-9. PMID 21706233. S2CID 8069716.
- ^ Amatu A, Sartore-Bianchi A, Moutinho C, Belotti A, Bencardino K, Chirico G, et al. (April 2013). «Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer». Clinical Cancer Research. 19 (8): 2265–72. doi:10.1158/1078-0432.CCR-12-3518. PMID 23422094.
- ^ Mokarram P, Zamani M, Kavousipour S, Naghibalhossaini F, Irajie C, Moradi Sarabi M, Hosseini SV (May 2013). «Different patterns of DNA methylation of the two distinct O6-methylguanine-DNA methyltransferase (O6-MGMT) promoter regions in colorectal cancer». Molecular Biology Reports. 40 (5): 3851–57. doi:10.1007/s11033-012-2465-3. PMID 23271133. S2CID 18733871.
- ^ Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, et al. (May 2005). «Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer». Gastroenterology. 128 (5): 1160–71. doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- ^ Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, et al. (April 2010). «Modulation of mismatch repair and genomic stability by miR-155». Proceedings of the National Academy of Sciences of the United States of America. 107 (15): 6982–87. Bibcode:2010PNAS..107.6982V. doi:10.1073/pnas.1002472107. PMC 2872463. PMID 20351277.
- ^ Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, et al. (April 2012). «Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer». Genome Integrity. 3 (1): 3. doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- ^ Human DNA Repair Genes, 15 April 2014, MD Anderson Cancer Center, University of Texas
- ^ Jin B, Robertson KD (2013). «DNA methyltransferases, DNA damage repair, and cancer». Advances in Experimental Medicine and Biology. 754: 3–29. doi:10.1007/978-1-4419-9967-2_1. ISBN 978-1-4419-9966-5. PMC 3707278. PMID 22956494.
- ^ Krishnan K, Steptoe AL, Martin HC, Wani S, Nones K, Waddell N, et al. (February 2013). «MicroRNA-182-5p targets a network of genes involved in DNA repair». RNA. 19 (2): 230–42. doi:10.1261/rna.034926.112. PMC 3543090. PMID 23249749.
- ^ Chaisaingmongkol J, Popanda O, Warta R, Dyckhoff G, Herpel E, Geiselhart L, et al. (December 2012). «Epigenetic screen of human DNA repair genes identifies aberrant promoter methylation of NEIL1 in head and neck squamous cell carcinoma». Oncogene. 31 (49): 5108–16. doi:10.1038/onc.2011.660. PMID 22286769.
- ^ Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, et al. (November 2008). «Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers». Molecular Cancer Research. 6 (11): 1710–17. doi:10.1158/1541-7786.MCR-08-0269. PMC 2948671. PMID 19010819.
- ^ Lam JS, Seligson DB, Yu H, Li A, Eeva M, Pantuck AJ, et al. (August 2006). «Flap endonuclease 1 is overexpressed in prostate cancer and is associated with a high Gleason score». BJU International. 98 (2): 445–51. doi:10.1111/j.1464-410X.2006.06224.x. PMID 16879693. S2CID 22165252.
- ^ Kim JM, Sohn HY, Yoon SY, Oh JH, Yang JO, Kim JH, et al. (January 2005). «Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells». Clinical Cancer Research. 11 (2 Pt 1): 473–82. doi:10.1158/1078-0432.473.11.2. PMID 15701830.
- ^ Wang K, Xie C, Chen D (May 2014). «Flap endonuclease 1 is a promising candidate biomarker in gastric cancer and is involved in cell proliferation and apoptosis». International Journal of Molecular Medicine. 33 (5): 1268–74. doi:10.3892/ijmm.2014.1682. PMID 24590400.
- ^ Krause A, Combaret V, Iacono I, Lacroix B, Compagnon C, Bergeron C, et al. (July 2005). «Genome-wide analysis of gene expression in neuroblastomas detected by mass screening» (PDF). Cancer Letters. 225 (1): 111–20. doi:10.1016/j.canlet.2004.10.035. PMID 15922863. S2CID 44644467.
- ^ Iacobuzio-Donahue CA, Maitra A, Olsen M, Lowe AW, van Heek NT, Rosty C, et al. (April 2003). «Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays». The American Journal of Pathology. 162 (4): 1151–62. doi:10.1016/S0002-9440(10)63911-9. PMC 1851213. PMID 12651607.
- ^ Sato M, Girard L, Sekine I, Sunaga N, Ramirez RD, Kamibayashi C, Minna JD (October 2003). «Increased expression and no mutation of the Flap endonuclease (FEN1) gene in human lung cancer». Oncogene. 22 (46): 7243–46. doi:10.1038/sj.onc.1206977. PMID 14562054.
- ^ Bi FF, Li D, Yang Q (2013). «Hypomethylation of ETS transcription factor binding sites and upregulation of PARP1 expression in endometrial cancer». BioMed Research International. 2013: 946268. doi:10.1155/2013/946268. PMC 3666359. PMID 23762867.
- ^ Bi FF, Li D, Yang Q (February 2013). «Promoter hypomethylation, especially around the E26 transformation-specific motif, and increased expression of poly (ADP-ribose) polymerase 1 in BRCA-mutated serous ovarian cancer». BMC Cancer. 13: 90. doi:10.1186/1471-2407-13-90. PMC 3599366. PMID 23442605.
- ^ Supek F, Lehner B (May 2015). «Differential DNA mismatch repair underlies mutation rate variation across the human genome». Nature. 521 (7550): 81–84. Bibcode:2015Natur.521…81S. doi:10.1038/nature14173. PMC 4425546. PMID 25707793.
- ^ a b Zheng CL, Wang NJ, Chung J, Moslehi H, Sanborn JZ, Hur JS, et al. (November 2014). «Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes». Cell Reports. 9 (4): 1228–34. doi:10.1016/j.celrep.2014.10.031. PMC 4254608. PMID 25456125.
- ^ Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM (April 2013). «The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα». Cell. 153 (3): 590–600. doi:10.1016/j.cell.2013.03.025. PMC 3641580. PMID 23622243.
- ^ Supek F, Lehner B (July 2017). «Clustered Mutation Signatures Reveal that Error-Prone DNA Repair Targets Mutations to Active Genes». Cell. 170 (3): 534–547.e23. doi:10.1016/j.cell.2017.07.003. hdl:10230/35343. PMID 28753428.
- ^ Polak P, Lawrence MS, Haugen E, Stoletzki N, Stojanov P, Thurman RE, et al. (January 2014). «Reduced local mutation density in regulatory DNA of cancer genomes is linked to DNA repair». Nature Biotechnology. 32 (1): 71–75. doi:10.1038/nbt.2778. PMC 4116484. PMID 24336318.
- ^ Swenberg JA, Lu K, Moeller BC, Gao L, Upton PB, Nakamura J, Starr TB (March 2011). «Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment». Toxicol Sci. 120 Suppl 1 (Suppl 1): S130–45. doi:10.1093/toxsci/kfq371. PMC 3043087. PMID 21163908.
- ^ a b Hamilton ML, Guo Z, Fuller CD, Van Remmen H, Ward WF, Austad SN, Troyer DA, Thompson I, Richardson A (May 2001). «A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA». Nucleic Acids Res. 29 (10): 2117–26. doi:10.1093/nar/29.10.2117. PMC 55450. PMID 11353081.
- ^ Ming X, Matter B, Song M, Veliath E, Shanley R, Jones R, Tretyakova N (March 2014). «Mapping structurally defined guanine oxidation products along DNA duplexes: influence of local sequence context and endogenous cytosine methylation». J Am Chem Soc. 136 (11): 4223–35. doi:10.1021/ja411636j. PMC 3985951. PMID 24571128.
- ^ a b c Zhou X, Zhuang Z, Wang W, He L, Wu H, Cao Y, Pan F, Zhao J, Hu Z, Sekhar C, Guo Z (September 2016). «OGG1 is essential in oxidative stress-induced DNA demethylation». Cell Signal. 28 (9): 1163–1171. doi:10.1016/j.cellsig.2016.05.021. PMID 27251462.
- ^ Poetsch AR (2020). «The genomics of oxidative DNA damage, repair, and resulting mutagenesis». Comput Struct Biotechnol J. 18: 207–219. doi:10.1016/j.csbj.2019.12.013. PMC 6974700. PMID 31993111.
- ^ D’Augustin O, Huet S, Campalans A, Radicella JP (November 2020). «Lost in the Crowd: How Does Human 8-Oxoguanine DNA Glycosylase 1 (OGG1) Find 8-Oxoguanine in the Genome?». Int J Mol Sci. 21 (21): 8360. doi:10.3390/ijms21218360. PMC 7664663. PMID 33171795.
- ^ Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A (September 2004). «In situ analysis of repair processes for oxidative DNA damage in mammalian cells». Proc Natl Acad Sci U S A. 101 (38): 13738–43. Bibcode:2004PNAS..10113738L. doi:10.1073/pnas.0406048101. PMC 518826. PMID 15365186.
- ^ Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, Costello JF, Wilkinson MF, Joung JK (December 2013). «Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins». Nat. Biotechnol. 31 (12): 1137–42. doi:10.1038/nbt.2726. PMC 3858462. PMID 24108092.
- ^ Ding N, Bonham EM, Hannon BE, Amick TR, Baylin SB, O’Hagan HM (June 2016). «Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage». J Mol Cell Biol. 8 (3): 244–54. doi:10.1093/jmcb/mjv050. PMC 4937888. PMID 26186941.
- ^ a b Jiang Z, Lai Y, Beaver JM, Tsegay PS, Zhao ML, Horton JK, Zamora M, Rein HL, Miralles F, Shaver M, Hutcheson JD, Agoulnik I, Wilson SH, Liu Y (January 2020). «Oxidative DNA Damage Modulates DNA Methylation Pattern in Human Breast Cancer 1 (BRCA1) Gene via the Crosstalk between DNA Polymerase β and a de novo DNA Methyltransferase». Cells. 9 (1): 225. doi:10.3390/cells9010225. PMC 7016758. PMID 31963223.
- ^ Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H (June 2005). «Recruitment of DNA methyltransferase I to DNA repair sites». Proc Natl Acad Sci U S A. 102 (25): 8905–9. Bibcode:2005PNAS..102.8905M. doi:10.1073/pnas.0501034102. PMC 1157029. PMID 15956212.
- ^ a b Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, Messina S, Iuliano R, Fusco A, Santillo MR, Muller MT, Chiariotti L, Gottesman ME, Avvedimento EV (July 2007). «DNA damage, homology-directed repair, and DNA methylation». PLOS Genet. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- ^ a b O’Hagan HM, Mohammad HP, Baylin SB (August 2008). «Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island». PLOS Genet. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- ^ Ha K, Lee GE, Palii SS, Brown KD, Takeda Y, Liu K, Bhalla KN, Robertson KD (January 2011). «Rapid and transient recruitment of DNMT1 to DNA double-strand breaks is mediated by its interaction with multiple components of the DNA damage response machinery». Hum Mol Genet. 20 (1): 126–40. doi:10.1093/hmg/ddq451. PMC 3000680. PMID 20940144.
- ^ Russo G, Landi R, Pezone A, Morano A, Zuchegna C, Romano A, Muller MT, Gottesman ME, Porcellini A, Avvedimento EV (September 2016). «DNA damage and Repair Modify DNA methylation and Chromatin Domain of the Targeted Locus: Mechanism of allele methylation polymorphism». Sci Rep. 6: 33222. Bibcode:2016NatSR…633222R. doi:10.1038/srep33222. PMC 5024116. PMID 27629060.
- ^ Farris MH, Texter PA, Mora AA, Wiles MV, Mac Garrigle EF, Klaus SA, Rosfjord K (December 2020). «Detection of CRISPR-mediated genome modifications through altered methylation patterns of CpG islands». BMC Genomics. 21 (1): 856. doi:10.1186/s12864-020-07233-2. PMC 7709351. PMID 33267773.
- ^ Allen B, Pezone A, Porcellini A, Muller MT, Masternak MM (June 2017). «Non-homologous end joining induced alterations in DNA methylation: A source of permanent epigenetic change». Oncotarget. 8 (25): 40359–40372. doi:10.18632/oncotarget.16122. PMC 5522286. PMID 28423717.
- ^ Cromie GA, Connelly JC, Leach DR (December 2001). «Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans». Molecular Cell. 8 (6): 1163–74. doi:10.1016/S1097-2765(01)00419-1. PMID 11779493.
- ^ O’Brien PJ (February 2006). «Catalytic promiscuity and the divergent evolution of DNA repair enzymes». Chemical Reviews. 106 (2): 720–52. doi:10.1021/cr040481v. PMID 16464022.
- ^ Maresca B, Schwartz JH (January 2006). «Sudden origins: a general mechanism of evolution based on stress protein concentration and rapid environmental change». The Anatomical Record Part B: The New Anatomist. 289 (1): 38–46. doi:10.1002/ar.b.20089. PMID 16437551.
- ^ «CRISPR gene-editing tool has scientists thrilled – but nervous» CBC news. Author Kelly Crowe. 30 November 2015.
External links[edit]
![]()
This audio file was created from a revision of this article dated 17 June 2005, and does not reflect subsequent edits.
- Media related to DNA repair at Wikimedia Commons
- Roswell Park Cancer Institute DNA Repair Lectures
- A comprehensive list of Human DNA Repair Genes
- 3D structures of some DNA repair enzymes
- Human DNA repair diseases
- DNA repair special interest group
- DNA Repair Archived 12 February 2018 at the Wayback Machine
- DNA Damage and DNA Repair
- Segmental Progeria
- DNA-damage repair; the good, the bad, and the ugly
ЧАСТЬ III. ПУТИ ПЕРЕДАЧИ ИНФОРМАЦИИ
Скучных ферментов не бывает.
— Артур Корнберг, «С любовью к ферментам», 1975
25. МЕТАБОЛИЗМ ДНК
В качестве хранилища генетической информации ДНК занимает центральное место среди биологических макромолекул. Нуклеотидная последовательность ДНК кодирует первичную структуру всех молекул клеточной РНК и белков и через ферменты опосредованно влияет на синтез всех других клеточных компонентов. Переход информации от ДНК к РНК и белку определяет размер, форму и жизнедеятельность всех живых существ.
ДНК прекрасно обеспечивает стабильное хранение генетической информации. Однако понятие «стабильное хранение» означает статичную неизменную картину. Оно не отражает сложности процессов, с помощью которых генетическая информация сохраняется и передается от одного поколения клеток к следующему. Метаболизм ДНК включает как процессы точного копирования молекул (репликация), так и процессы, которые влияют на саму структуру, несущую информацию (репарация и рекомбинация). Именно эти процессы мы и рассмотрим в данной главе.
Метаболизм ДНК должен происходить идеально точно. Химические реакции присоединения одного нуклеотида к другому при репликации ДНК элегантны и на удивление просты. Сложности, как мы увидим далее, в ферментативном аппарате, обеспечивающем точность передачи генетической информации. Ошибки, возникающие при синтезе ДНК, могут привести к серьезным последствиям, причем не только потому, что они изменяют или нарушают функцию определенного гена, но и потому, что такое изменение передается по наследству.
Ферменты, участвующие в синтезе ДНК. копируют молекулы ДНК, содержащие миллионы оснований. Они проделывают это с необыкновенной точностью и скоростью, несмотря на то что ДНК очень компактно упакована и связана с другими белками. Образование фосфодиэфирных связей между нуклеотидами в растущей цепи ДНК — только часть сложного процесса, в котором задействовано множество белков и ферментов.
Задача сохранения генетической информации лежит в основе процесса репарации ДНК. В гл. 8 (т. 1) подробно описана чувствительность ДНК ко многим разрушительным воздействиям. Такие воздействия происходят нечасто, но они играют очень важную роль, поскольку устойчивость организмов к изменениям последовательности ДНК очень низкая. ДНК — единственная макромолекула, для которой предусмотрена система репарации; количество, разнообразие и сложность механизмов репарации отражает многообразие пагубных для ДНК воздействий.
Клетки могут перестраивать хранящуюся в них генетическую информацию в процессах под общим названием «рекомбинация». Казалось бы, процесс рекомбинации подрывает принцип первостепенного значения стабильности и целостности генетической информации. Однако на самом деле большинство перестроек ДНК играет конструктивную роль в поддержании целостности генома, специфическим образом влияя на репликацию и репарацию ДНК и расхождение хромосом.
В этой главе особое внимание уделяется ферментам метаболизма ДНК. Они заслуживают внимательного изучения не только ввиду их большого биологического значения и чисто научного интереса, но и в связи с их возрастающей ролью в качестве лекарственных препаратов в медицине и в качестве инструментов в широком круге современных биохимических технологий. Многие ценные открытия в области метаболизма ДНК были сделаны на клетках Escherichia coli, поэтому для объяснения основ метаболизма обычно в качестве примера служат хорошо известные ферменты этой бактерии. Беглый взгляд на некоторые важные гены на генетической карте Е. coli (рис. 25-1) позволяет представить себе сложность ферментных систем, участвующих в метаболизме ДНК.
Рис. 25-1. Хромосомная карта Escherichia coli. Показано расположение генов, кодирующих многие важные для метаболизма ДНК белки. Количество известных генов, участвующих в метаболизме ДНК, свидетельствует о сложности этих процессов. Числа от 0 до 100 внутри кольцевой хромосомы соответствуют генетическим единицам измерения, называемым минутами. Каждая минута соответствует отрезку молекулы ДНК длиной примерно 40 000 п. н. Трехбуквеные названия генов обычно отражают некоторые аспекты их функции, к примеру, mut — мутагенез; dna — репликация ДНК; pol — ДНК-полимераза; rро — РНК-полимераза; uvr — устойчивость к УФ; rес — рекомбинация; dam — метилирование аденина; lig — ДНК-лигаза; Теr — терминация репликации; ori — точка начала репликации (у Е. coli это oriC, как на рисунке).

Прежде чем перейти к детальному изучению процесса репликации, сделаем короткое отступление и поговорим о принятых сокращениях названий бактериальных генов и белков, поскольку со многими из них нам предстоит встретиться в этой и последующих главах. Аналогичные договоренности используются и в обозначениях эукариотических генов, хотя конкретное сокращение может зависеть от вида организма, и единого правила для всех эукариотических систем не существует.
Ключевые договоренности.
Бактериальные гены обычно обозначают тремя строчными буквами курсивом, и, как правило, название отражает функцию этих генов. Например, dna, uvr и rес гены означают репликацию ДНК, устойчивость к поражающему действию УФ-облучения и рекомбинацию соответственно. Если несколько генов отвечают за одну и ту же функцию, они дополнительно обозначаются буквами А, В, С и т. д., например, dnaA, dnaB, dnaQ, что обычно указывает на порядок их открытия, а не порядок участия в цепи реакций. ■
Использование сокращений в названиях белков менее очевидно. В ходе генетических исследований обычно выделяют и характеризуют белковые продукты каждого гена. Многие бактериальные гены были идентифицированы и названы до того, как выяснилось значение их белковых продуктов. Иногда оказывается, что продукт гена — это уже известный белок, и его приходится переименовывать. Однако часто бывает, что продукт гена еще неизвестен и обладает активностью, которую нельзя описать обычным названием фермента.
Ключевые договоренности.
Бактериальные белки часто сохраняют названия соответствующих генов. Названия белков Е. coli пишутся прямым шрифтом с заглавной буквы, например, белковые продукты генов dnaA и rесА называются DnaA и RecA соответственно. ■
25.1. Репликация ДНК
Задолго до установления структуры ДНК ученых удивляла способность организмов воссоздавать самих себя и способность клеток образовывать много идентичных копий крупных и сложных макромолекул. Рассуждения на эту тему концентрировались вокруг концепции матрицы — структуры, которая позволяет молекулам соединяться в определенном порядке и образовывать макромолекулу с уникальной последовательностью и функцией. В 1940-е гг. уже сложилось представление о том, что носителем генетической информации является ДНК, но только когда Джеймс Уотсон и Фрэнсис Крик установили ее структуру, стало понятно, каким образом ДНК играет роль матрицы для репликации и передает генетическую информацию: дело в том, что одна ее цепь комплементарна другой. Основания образуют пары по определенным правилам, и каждая цепь является матрицей для новой цепи с предсказуемой комплементарной последовательностью (см. рис. 8-14, 8-15 в т. 1).
Было доказано, что основные свойства процесса репликации ДНК и каталитические механизмы этого процесса в значительной степени идентичны у всех видов организмов. Именно это единство механизмов мы постараемся подчеркнуть в нашем обсуждении, продвигаясь от общих основ процесса репликации к ферментам репликации Е. coli и, наконец, к репликации эукариот.
Основные принципы репликации ДНК
В ранних исследованиях репликации бактериальной ДНК и ее ферментов было установлено несколько базовых принципов, на которых основан синтез ДНК у всех живых существ.
Репликация ДНК полуконсервативна.
Каждая цепь ДНК служит матрицей для синтеза новой цепи, при этом образуются две новые двухцепочечные молекулы ДНК, каждая из которых состоит из одной новой и одной старой цепей. Поэтому процесс называется полуконсервативной репликацией.
Уотсон и Крик выдвинули гипотезу полуконсервативной репликации вскоре после публикации своей статьи о структуре ДНК с 1953 г.; в 1957 г. эта гипотеза была подтверждена Мэтью Мезельсоном и Франклином Сталем, которые искусно провели следующие эксперименты. Мезельсон и Сталь культивировали клетки Е. coli на протяжении многих генераций в среде, в которой единственный источник азота (NH4Cl) содержал тяжелый изотоп азота 15N вместо наиболее распространенного легкого изотопа 14N. Плотность выделенной из этих клеток ДНК примерно на 1% больше, чем у обычной [14N] ДНК (рис. 25-2, а). Несмотря на столь небольшое различие, смесь тяжелой [15N] ДНК и легкой [14N] ДНК можно разделить центрифугированием в градиенте плотности хлорида цезия.
Рис. 25-2, а — несколько поколений клеток выращивали в среде, содержащей только тяжелый азот 15N, поэтому в ДНК этих клеток содержался исключительно азот 15N, что показано в виде одной (синей) полосы, образующейся при центрифугировании в градиенте плотности CsCl. б — клетки переносили на среду, содержащую только легкий азот 14N, и после первого деления клеток выделяли ДНК, которая в градиенте хлорида цезия образовывала более высокую (фиолетовую) полосу, в — после второго цикла репликации ДНК разделялась на две полосы: гибридную (фиолетовую) и еще более легкую (красную), содержащую только ДНК с 14N, что подтверждало полу- консервативный характер репликации.

Клетки Е. coli, выращенные на среде с 15N, переносили на свежую питательную среду, содержащую только изотоп 14N, где клетки продолжали расти до тех пор, пока численность популяции не удваивалась. ДНК, выделенную из этой первой генерации клеток, центрифугировали в градиенте CsCl и наблюдали единственную полосу, положение которой свидетельствовало о том, что двухспиральная ДНК дочерних клеток представляет собой гибрид, содержащий одну новую цепь с 14N и одну родительскую цепь с 15N (рис. 25-2, б).
Представленные результаты опровергли альтернативную теорию консервативной репликации, согласно которой одна дочерняя молекула ДНК содержит две вновь синтезированные цепи, а другая дочерняя молекула — две родительские цепи; если бы эта теория была верна, в эксперименте Мезельсона -Сталя не было бы гибридной ДНК. Гипотеза полуконсервативной репликации дополнительно подтверждалась на следующем этапе эксперимента (рис. 25-2, в). После второго деления и удвоения клеток на среде с 14N из них была выделена ДНК, разделившаяся в градиенте хлорида цезия на две полосы: одна по плотности соответствовала легкой ДНК, а другая — гибридной ДНК. полученной после первого деления клеток.
Репликация начинается в точке начала репликации и обычно идет в двух направлениях.
После доказательства полуконсервативного механизма репликации возникло множество вопросов. Полностью ли раскручивается родительская ДНК до начала репликации каждой цепи? Начинается ли репликация в случайной или в какой-то особой точке? После инициации в какой-то точке репликация происходит в одном направлении или в двух?
В ранних исследованиях Джона Кэрнса, проведенным с помощью метода авторадиографии, удалось доказать, что репликация — высококоординированный процесс, в котором родительские цепи одновременно расплетаются и реплицируются. Кэрнс получил радиоактивную ДНК Е. coli, для чего культивировал эту бактерию на среде с меченным тритием (3Н) тимидином. ДНК была аккуратно выделена, покрыта слоем фотографической эмульсии и оставлена так на несколько недель; за это время радиоактивный тимидин оставил в фотоэмульсии «следы» — зерна серебра, создав фотоотпечаток молекулы ДНК. По этим отпечаткам видно, что интактная хромосома Е. coli представляет собой единую гигантскую кольцевую структуру длиной 1,7 мм. В радиоактивной ДНК, выделенной из клеток во время репликации, кроме того, была обнаружена дополнительная петля (рис. 25-3, а). Кэрнс сделал вывод, что, петля образуется в результате формирования двух радиоактивных дочерних цепей, каждая из которых комплементарна родительской пени. На одном или на обоих концах петли движется репликативная вилка, в которой родительская ДНК раскручивается, позволяя реплицироваться разделившимся цепям. Согласно полученным Кэрнсом данным, обе цепи ДНК реплицируются одновременно, а различные варианты его эксперимента (рис. 25-3, б) показали, что репликация бактериальных хромосом двунаправленная: на обоих концах петли расположены активные репликативные вилки.
Рис. 25-3. Выявление двунаправленного характера репликации ДНК. а — во время репликации кольцевой хромосомы образуется структура, напоминающая греческую букву тета (θ), так как обе нити реплицируются одновременно (новая нить показана красным цветом). б — репликация может происходить в одном или в обоих направлениях, что можно установить с помощью авторадиографии: при внесении 3Н на короткий период непосредственно перед прекращением реакции метка (отмечена красным) обнаруживается в одной или в двух репликативных вилках соответственно. Так была продемонстрирована двунаправленность репликации у Е. coli, Bacillus subtilis и других бактерий. На авторадиограмме показан репликационный «глазок» в ДНК В. subtilis. Самая большая плотность зерен серебра (стрелки) наблюдается именно в тех двух местах, где происходит репликация. Не подвергшаяся репликации часть хромосомы, находящаяся вне глазка, не содержит метки и поэтому невидима.

Чтобы выяснить, образуются ли репликативные вилки в каком-то определенном участке ДНК, необходимо было установить по всей длине молекулы некие реперные точки. Это было сделано с помощью метода денатурирующего картирования, разработанного Россом Инманом с коллегами. С помощью хромосомы бактериофага λ длиной 48 502 п. и. было показано, что можно вызвать избирательную денатурирацию ДНК в последовательностях с необычно высоким содержанием пар А = Т, получая воспроизводимую картину одноцепочечных глазков (см. рис. 8-28 в т. 1). Таким же образом можно частично денатурировать выделенную ДНК, содержащую репликативные петли. Это позволяет измерить и картировать положение и движение репликативных вилок, используя денатурированные области в качестве реперных точек. С помощью данного метода было показано, что в этой системе репликативные петли всегда возникают в особых точках, которые были названы точками начала репликации (ориджинами). Получил подтверждение и выявленный ранее двунаправленный характер репликации. У кольцевых молекул ДНК две репликативные вилки встречаются в точке, расположенной напротив точки начала репликации. Специфические точки начала репликации с тех пор были идентифицированы и охарактеризованы у бактерий и низших эукариот.
Синтез ДНК наполовину прерывистый и проходит в направлении 5’—> 3′.
Новая цепь ДНК всегда синтезируется в направлении 5′ —> 3′, т. е. приращение ДНК происходит со стороны свободной ОН-группы на 3′-конце (строение 5′- и 3′-концов цепи ДНК см. на рис. 8-7 в т. 1). Поскольку две цепи ДНК антипараллельны, цепь, служащая в качестве матрицы, считывается с 3’-конца в направлении к 5′-концу.
Но если синтез ДНК всегда происходит в направлении 5′ —> 3′, как могут обе цепи синтезироваться одновременно? Если обе цепи синтезируются непрерывно, пока движется репликативная вилка, одна цепь должна синтезироваться в направлении 3′ —> 5′. Эту проблему в 1960-х гг. разрешил Рейджи Оказаки с коллегами. Они обнаружили, что одна из новых цепей ДНК синтезируется в виде коротких отрезков, которые теперь называются фрагментами Оказаки. В результате их исследований стало ясно, что одна цепь синтезируется непрерывно, а вторая — прерывисто (рис. 25-4). Направление синтеза непрерывной, или лидирующей, цепи совпадает с направлением движения репликативной вилки. Направление синтеза прерывистой, или отстающей, цепи противоположно направлению движения вилки. Фрагменты Оказаки различаются по длине от нескольких сотен до нескольких тысяч нуклеотидов в зависимости от типа клеток. Ниже мы увидим, что синтез лидирующей и отстающей цепей четко координируется.
Рис. 25-4. Различия цепей ДНК на репликативной вилке. Новая цепь ДНК (красная) всегда синтезируется в направлении 5′ —> 3′. Матрица считывается в противоположном направлении. 3′ —> 5′. Лидирующая цепь непрерывно синтезируется по направлению движения репликативной вилки. Другая цепь, отстающая, синтезируется прерывисто в виде коротких отрезков (фрагментов Оказаки) в направлении, противоположном тому, в котором движется репликативная вилка. Фрагменты Оказаки сшиваются ДНК-лигазой. У бактерий длина фрагментов Оказаки составляет примерно от 1000 до 2000 нуклеотидов. В клетках эукариот они короче: от 150 до 200 нуклеотидов.

ДНК разрушается нуклеазами
Чтобы объяснить энзимологию процесса репликации ДНК, сначала рассмотрим ферменты, которые разрушают, а не синтезируют ДНК. Эти ферменты называют нуклеазами или ДНКазами, если они обладают большей специфичностью к ДНК, чем к РНК. Каждая клетка содержит несколько разных нуклеаз, принадлежащих к двум большим классам: экзонуклеазы и эндонуклеазы. Экзонуклеазы расщепляют нуклеиновые кислоты с одного конца молекулы. Многие из них работают только в направлении 5′ —> 3′ или только в направлении 3′ —> 5′, удаляя нуклеотиды либо с 5′-конца, либо с 3′-конца одной цепи двухцепочечной молекулы нуклеиновой кислоты или одноцепочечной ДНК. Эндонуклеазы начинают расщепление нуклеиновой кислоты со специфических внутренних сайтов, расщепляя цепь на более мелкие фрагменты. Некоторые экзонуклеазы и эндонуклеазы расщепляют только одноцепочечные ДНК. Существует несколько важных классов эндонуклеаз, которые разрушают только определенные нуклеотидные последовательности (например, эндонуклеазы рестрикции, которые имеют очень большое значение в биотехнологии; см. гл. 9, рис. 9-2 вт. 1). В этой и в последующих главах мы встретим много типов нуклеаз.
ДНК синтезируется ДНК-полимеразами
Поиск фермента, который может синтезировать ДНК, начался в 1955 г. Артуру Корнбергу с коллегами удалось очистить и охарактеризовать ДНК-полимеразу из клеток Е. coli. Этот фермент, состоящий из одного полипептида, теперь называют ДНК-полимеразой I (Мr = 103 000; кодируется геном роlА). Гораздо позже исследователи обнаружили, что Е. coli содержит еще как минимум четыре другие ДНК-полимеразы, описанные ниже.

При детальном изучении ДНК-полимеразы 1 были установлены общие для всех ДНК-полимераз характеристики процесса синтеза ДНК. В основной реакции происходит перенос фосфорильных групп. При этом 3′-гидроксильная группа нуклеотида на 3′-конце растущей цепи выступает в роли нуклеофила, атакующего α-фосфор присоединяющегося дезоксинуклеозид-5′-трифосфата (рис. 25-5). В результате высвобождается неорганический пирофосфат. Основная реакция выглядит следующим образом:

где dNMP и dNTP — дезоксинуклеозид-5v-монофосфат и дезоксинуклеозид-5′-трифосфат соответственно. Реакция происходит с минимальным изменением свободной энергии, поскольку одна фосфодиэфирная связь образуется из менее стабильного фосфоангидрида. Нековалентные стэкинговые взаимодействия оснований и их спаривание обеспечивают дополнительную стабилизацию удлиняющейся ДНК по сравнению со свободными нуклеотидами. Кроме того, синтез ДНК приводит к высвобождению в клетке 19 кДж/моль энергии в результате последующего гидролиза пирофосфата под действием фермента пирофосфатазы (с. 38 в т. 2).
Рис. 25-5. МЕХАНИЗМ РЕАКЦИИ. Элонгация цепи ДНК. а — ДНК-полимераза I нуждается в одной неспаренной цепи в качестве матрицы и в праймере, несущем на 3′-конце свободную гидроксильную группу, к которой присоединяется новое нуклеотидное звено. Каждый присоединяющийся нуклеотид выбирается по принципу комплементарности по отношению к соответствующему нуклеотиду в матричной цепи. Продукт реакции вновь содержит свободную 3′-гидроксильную группу, позволяющую присоединять следующий нуклеотид. б — для катализа, вероятно, требуется два иона Мg2+, координированных с двумя фосфатными группами присоединяющегося нуклеозидтрифосфата и с тремя остатками Asp, два из которых консервативны у всех ДНК-полимераз. Ион Мg2+, изображенный справа, облегчает атаку 3′-гидроксильной группы праймера на α-фосфат нуклеозидтрифосфата другой ион Мg2+ способствует удалению пирофосфата. Оба иона стабилизируют структуру пятикоординационного переходного состояния. По схожему механизму действует и РНК-полимераза (см. рис. 26-1, б).

Еще в ранних исследованиях ДНК-полимеразы I удалось определить два основных условия, необходимых для полимеризации ДНК. Во-первых, все ДНК-полимеразы нуждаются в матрице. Реакция полимеризации происходит на матрице ДНК согласно правилам комплементарности оснований, установленным Уотсоном и Криком: если в матрице присутствует гуанин, к новой цепи присоединяется дезоксинуклеотид цитозина и т. д. Это открытие имеет очень важное значение не только потому, что оно объясняет химическую основу точной полуконсервативной репликации ДНК, но и потому, что это был первый пример использования матрицы для проведения реакции биосинтеза.
Во-вторых, полимеразы нуждаются в праймере. Праймер — это участок цепи (комплементарный матрице) со свободной З’-гидроксильной группой, к которой может присоединиться нуклеотид. Иначе говоря, перед началом синтеза часть новой цепи уже должна существовать: все ДНК-полимеразы могут присоединять нуклеотиды только к уже существующей цепи. Многие праймеры представляют собой олигонуклеотиды РНК, а не ДНК; специальные ферменты синтезируют праймеры там и тогда, где и когда они требуются.
После присоединения нуклеотида к растущей цепи ДНК ДНК-полимераза либо диссоциирует, либо продвигается вдоль матрицы и присоединяет следующий нуклеотид. Диссоциация и обратное присоединение полимеразы может ограничивать среднюю скорость полимеризации — процесс обычно протекает быстрее, если полимераза присоединяет нуклеотиды, не покидая матрицы. Среднее число нуклеотидов, присоединенных до диссоциации полимеразы, определяет ее процессивность. ДНК-полимеразы существенно различаются по процессивности; некоторые присоединяют всего несколько нуклеотидов до диссоциации, другие присоединяют многие тысячи.
Репликация — очень точный процесс
Репликация осуществляется с исключительной точностью. У Е. coli происходит всего одна ошибка на каждые 109-1010 присоединенных нуклеотидов. Для хромосомы Е. coli, размер которой составляет примерно 4,6 • 106 п. н., это означает, что за 1000-10 000 циклов репликации случается лишь одна ошибка. Во время полимеризации выбор между правильным и неправильным нуклеотидом осуществляется не только по специфичности водородных связей между комплементарными основаниями в парах, но также по геометрии стандартных А = Т и G = C пар оснований (рис. 25-6). В активном центре ДНК-полимеразы I могут разместиться только пары оснований с правильной геометрией. Неподходящий нуклеотид может образовать водородные связи с нуклеотидом в матрице, но такая пара обычно не попадает в активный центр фермента. Это позволяет отбраковать неправильное основание до того, как сформируется фосфодиэфирная связь.
Рис. 25-6. Значение геометрии пар оснований для точности репликации ДНК. а — стандартные пары оснований А = Т и G = C очень схожи по геометрии, и, если активный центр фермента может вместить одну пару (синяя рамка), то обычно он вмещает и другую, б — геометрия аномальных пар отличается, и они не помещаются в активном центре ДНК-полимеразы I.

Однако, чтобы обеспечить высокую степень точности репликации, одной только точности реакции полимеризации недостаточно. Измерения in vitro показали, что ДНК-полимераза вставляет один неправильный нуклеотид на 104 — 105 правильных нуклеотидов. Иногда эти ошибки возникают из-за того, что основание кратковременно находится в нехарактерной таутомерной форме (см. рис. 8-9 в т. 1), что позволяет ему образовать водородные связи не с тем партнером. Число ошибок in vivo уменьшается благодаря дополнительным ферментативным механизмам.
Один механизм сокращения числа ошибок свойствен практически всем ДНК- полимеразами и заключается в независимой 3′ —> 5′-экзонуклеазной активности, благодаря которой каждый нуклеотид после присоединения проверяется дважды. Эта нуклеазная активность чрезвычайно чувствительна к аномальному спариванию оснований, что позволяет ферменту удалять ошибочно присоединенный нуклеотид (рис. 25-7). Если полимераза присоединила неправильный нуклеотид, происходит ингибирование ее перемещения к позиции, в которой должен присоединяться следующий нуклеотид. Эта пауза создает возможность для исправления ошибки. С помощью 3′ —> 5′-экзонуклеазной активности полимераза удаляет неправильный нуклеотид и продолжает удлинение цепи. Данная активность, называемая корректирующей, не просто обращение полимеразной реакции (уравнение 25-1), ведь в ней не участвует пирофосфат. Полимеразную и корректирующую активности ДНК-полимеразы можно измерить независимо. Корректирующая активность дополнительно увеличивает точность реакции полимеризации в 102-103раз. У мономерной ДНК-полимеразы I полимеразная и корректирующая активности локализованы в разных активных центрах в одном и том же полипептиде.
Рис. 25-7. Пример исправления ошибки благодаря 3′ —> 5′-экзонуклеазной активности ДНК-полимеразы I. При изучении структуры фермента было установлено, что при ориентации фермента по ходу движения ДНК- центр его экзонуклеазной активности локализован впереди центра полимеразной активности. Ошибочное спаривание (здесь ошибка С-А) препятствует транслокации ДНК-полимеразы I в следующее положение. Фермент скользит назад, исправляет ошибку благодаря своей 3′ —> 5′-экзонуклеазной активности, а затем возобновляет полимеразную активность в направлении 5′ —> 3′.

При тщательном подборе оснований в сочетании с исправлением ошибок ДНК-полимераза совершает примерно одну ошибку на 106-108 присоединенных оснований. В реальности измеряемая точность репликации Е. coli еще выше. Дополнительная точность обеспечивается независимой ферментативной системой, которая заменяет аномальные пары оснований, оставшиеся после репликации. Данный процесс исправления ошибок, наряду с другими процессами репарации ДНК, описан в разд. 25.2.
У Е. coli не менее пяти ДНК-полимераз
Более 90% ДНК-полимеразной активности в экстрактах Е. coli связано с функцией ДНК-полимеразы I. Однако вскоре после выделения этого фермента в 1955 г. стали накапливаться сведения о том, что он не может осуществлять репликацию крупной хромосомы Е. coli. Во-первых, скорость, с которой фермент присоединяет нуклеотиды (600 нуклеотидов/мин), не соответствует скорости движения репликативной вилки в бактериальной клетке (скорость движения репликативной вилки, как минимум, в 100 раз выше). Во- вторых, ДНК-полимераза I обладает относительно низкой процессивностью. В-третьих, генетические исследования показали, что в репликации участвуют многие гены и, следовательно, многие белки: очевидно, что ДНК-полимераза I действует не в одиночку. В-четвертых, и это особенно важно, в 1969 г. Джон Кэрнс выделил бактериальный штамм с поврежденным геном ДНК-полимеразы I, с которого синтезировался неактивный фермент. Хотя этот штамм был чрезвычайно чувствителен к повреждениям ДНК, тем не менее он был жизнеспособен!
Поиск других ДНК-полимераз привел к открытию в начале 1970-х гг. ДНК-полимеразы II и ДНК-полимеразы III у Е. coli. ДНК-полимераза II участвует в одном из механизмов репарации ДНК (разд. 25.3). ДНК-полимераза III — главный фермент репликации у Е. coli. Свойства трех ДНК-полимераз Е. coli сравниваются в табл. 25-1. ДНК-полимеразы IV и V, идентифицированные в 1999 г., принимают участие в необычном варианте репарации ДНК (разд. 25.2).
Таблица 25-1. Сопоставление ДНК-полимераз Е. coli
|
ДНК-полимер |
аза |
||
|
I |
II |
III |
|
|
Структурный гена |
polA |
роlB |
polC (dnaE) |
|
Субъединицы (количество разных типов) |
1 |
7 |
≥10 |
|
Относительная молекулярная масса (Mr) |
103 000 |
88 000б |
791 500 |
|
3′ —> 5′-экзонуклеаза (коррекция ошибок) |
Да |
Да |
Да |
|
5′ —> 3′-экзонуклеаза |
Да |
Нет |
Нет |
|
Скорость полимеризации (нуклеотиды/с) |
16-20 |
10 |
250- 1000 |
|
Процессивность (число нуклеотидов, присоединенных до диссоциации полимеразы) |
3-200 |
1500 |
≥500 000 |
а Для субъединичных ферментов назван ген, кодирующий субъединицу с полимеразной активностью. Ген dnаE в настоящее время называется polC.
б Только субъединица, отвечающая за полимеризацию. В составе ДНК-полимеразы II есть те же субъединицы, что и у ДНК-полимеразы III: (β-, y-, δ-, δ’-, χ- и Ψ-субъединицы (см. табл. 25-2).
ДНК-полимераза I, таким образом, не главный фермент в репликации; она выполняет функцию проверки и поддержания порядка во время репликации, рекомбинации и репарации. Особые функции полимеразы дополняются ее 5′ —> 3′-экзо- нуклеазной активностью. Эта активность, отличная от корректирующей 3′ —> 5′-экзонуклеазной активности (рис. 25-7), локализована в структурном домене, который может быть отщеплен от фермента в условиях мягкой обработкой протеазами. При удалении 5′ —> 3′-экзонуклеазного домена остающийся фрагмент (Мr = 68 000), называемый большим фрагментом, или фрагментом Клёнова (рис. 25-8), сохраняет полимеразную и корректирующую активность. Интактная ДНК-полимераза I благодаря своей 5′ —> 3′-экзонуклеазной активности в ходе так называемой ник-трансляции может заменить участок ДНК (или РНК), связанный с матричной цепью (рис. 25-9). Большинство других ДНК-полимераз не имеют 5′ —> 3′-экзонуклеазной активности.
Рис. 25-8. Большой фрагмент ДНК-полимеразы I (фрагмент Клёнова). Эта полимераза широко распространена у бактерий. Фрагмент Клёнова, образующийся в результате протеолитической обработки полимеразы, сохраняет полимеразную и корректирующую активности. Представленный на рисунке фрагмент Клёнова получен из термофильной бактерии Bacillus stearothermophilus (PDB ID 3BDP). Активный центр полимеразы находится глубоко в щели около дальнего конца связанной ДНК (синяя; темно-синим цветом изображена матричная цепь).

Рис. 25-9. Ник-трансляция. В ходе этого процесса участок РНК или ДНК, спаренный с матричной ДНК, одновременно разрушается под действием 5′ —> 3′-экзонуклеазной активности ДНК-полимеразы I и заменяется на новый под действием полимеразной активности того же фермента. Эта функция реализуется в процессе репарации ДНК и при удалении РНК-праймеров в ходе репликации (оба процесса описаны ниже). Цепь нуклеиновой кислоты (ДНК или РНК), подлежащая удалению, показана зеленым, новая цепь — красным. Синтез ДНК начинается с ника (разрыва фосфодиэфирной связи, освобождающего 3′-гидроксильную и 5′-фосфатную группы). Полимераза I удлиняет нематричную цепь ДНК и перемещает ник вдоль ДНК. Этот процесс называется ник-трансляцией. В месте диссоциации ДНК-полимеразы ник сохраняется и позднее зашивается другим ферментом.

ДНК-полимераза III устроена намного сложнее, чем ДНК-полимераза I; она состоит из субъединиц десяти разных типов (табл. 25-2). Ее полимеразная и корректирующая активности связаны с α- и ε-субъединицами соответственно. При объединении θ-субъединицы с α- и ε-субъединицами образуется корфермент, который уже может осуществлять синтез ДНК, но с ограниченной процессивностью. Два кор-фермента полимеразы могут связываться друг с другом посредством еще одного субъединичного комплекса, называемого погрузчиком зажима, или y-комплексом, состоящим из пяти субъединиц четырех разных типов Ƭ2yδδ’. Кор-ферменты связаны через Ƭ-субъединицы. Две дополнительные единицы χ и Ψ, присоединены к погрузчику зажима. Полный набор из 13 белковых субъединиц (девяти разных типов) называется ДНК-полимеразой III* (рис. 25-10, а).
Таблица 25-2. Субъединицы ДНК-полимеразы III E. coli

а y-Субъединицу кодирует часть гена Ƭ-субъединицы, так что 66% Ƭ-субъединицы с N-конца совпадают по аминокислотной последовательности с y-субъединицей. y-Субъединица образуется в результате смещения рамки считывания при трансляции (см. рис. 27-9), что приводит к преждевременной остановке трансляции.
ДНК-полимераза III* способна полимеризовать ДНК, но с намного меньшей процессивностью, чем нужно для организованной репликации целой хромосомы. Необходимое увеличение процессивности обеспечивается присоединением четырех β-субъединиц с образованием полного фермента ДНК-полимеразы III. β-Субъединицы объединяются в пары и формируют структуры в форме бубликов, которые охватывают молекулы ДНК, как зажимы (рис. 25-10, б). Каждый димер связывается с кор-ферментом полимеразы III* (один димерный зажим на кор-фермент) и во время репликации скользит вдоль ДНК. Скользящий β-зажим предупреждает диссоциацию ДНК-полимеразы III из комплекса с ДНК, резко увеличивая процессивность до 500 000 нуклеотидов и более (табл. 25-1).
Рис. 25-10. ДНК-полимераза III. а — строение бактериальной ДНК-полимеразы III. Два центральных домена (кор-ферменты), состоящие из субъединиц α, ε и θ, связаны с y-комплексом (погрузчиком зажима), состоящим из пяти субъединиц Ƭ2yδδ’. Субъединицы y и Ƭ кодируются одним и тем же геном. Субъединица у представляет собой укороченную субъединицу Ƭ: субъединица Ƭ содержит домен, идентичный у, и дополнительный сегмент, взаимодействующий с кор-ферментом. Две другие субъединицы χ и Ψ (не показаны) ДНК-полимеразы III* также связаны с y-комплексом. Два β-зажима присоединяются к двум кор-ферментам; каждый зажим — это димер β-субъединицы. Комплекс взаимодействует с DnaB- хеликазой через Ƭ-субъединицу. б — две β-субъединицы полимеразы III Е. coli образуют зажим в виде кольца, которое окружает ДНК. Зажим скользит по молекуле ДНК, увеличивая процессивность холофермента ДНК-полимеразы III до 500 000 нуклеотидов и более, не позволяя ДНК диссоциировать. Вид снизу демонстрирует две β-субъединицы, изображенные в виде лент голубого и серого цвета, окружающих СРК-модель молекулы ДНК. Вид сбоку демонстрирует контур поверхности β-субъединиц (серого цвета), которые окружают двойную спираль ДНК (голубой и синий цвет), изображенную в виде стрежневой модели (по PDB ID 2PОL).

В репликации ДНК участвует множество ферментов и белковых факторов
Для репликации Е. coli нужна не только ДНК-полимераза, но также не менее 20 различных ферментов и белков, каждый из которых выполняет определенную задачу. Весь этот комплекс был назван ДНК-репликативной системой, или реплисомой. Сложность ферментативного аппарата репликации связана со структурой ДНК и с требованиями высокой точности синтеза. Основные классы ферментов репликации перечислены ниже в соответствии с теми функциями, которые они выполняют.
Чтобы обеспечить доступ к цепям ДНК, которые служат в качестве матрицы, две родительские цепи нужно разделить. Обычно этот процесс осуществляют хеликазы — ферменты, которые движутся вдоль ДНК и разделяют ее цепи, затрачивая при этом энергию АТР. Разделение цепей создает топологическое напряжение в спиральной структуре ДНК (см. рис. 24-12), которое снимается топоизомеразами. Разделенные цепи стабилизируются ДНК-связывающими белками. Как уже было сказано, прежде чем ДНК-полимеразы смогут начать синтез ДНК, на матрице должны находиться праймеры — обычно это короткие сегменты РНК, синтезированные ферментами праймазами. Позднее РНК-праймеры удаляются и заменяются на ДНК; у Е. coli это действие — одна из основных функций ДНК-полимеразы I. После вырезания РНК-праймера и встраивания ДНК в последовательности ДНК сохраняется ник в форме разорванной фосфодиэфирной связи. Эти ники зашивают ДНК- лигазы. Все перечисленные процессы нуждаются в координации и регуляции, которые хорошо исследованы в системе Е. coli.
Репликация хромосомы E. coli происходит постадийно
В синтезе молекулы ДНК можно выделить три стадии: инициация, элонгация и терминация, которые различаются как по химической сути, так и по составу ферментов. В этой и двух последующих главах рассматриваются именно эти три общие стадии синтеза крупных информационных полимеров ДНК, РНК и белков, а также специфические особенности каждого биосинтетического пути. Описываемые далее события базируются на информации, первоначально полученной в экспериментах in vitro на очищенных белках Е. coli, хотя принципы репликации весьма консервативны во всех системах.
Инициация.
Точка начала репликации Е. coli (oriC) состоит из 245 п. н.; она содержит элементы последовательности, консервативные во многих бактериальных точках начала репликации. Стандартный набор консервативных последовательностей показан на рис. 25-11. Наибольший интерес представляют два типа последовательностей: пять повторов размером по 9 п. н. (R-сайты) — участки связывания ключевого инициаторного белка DnaA, а также А = Т-богатый участок, называемый ДНК-расплетающим элементом DUE (от англ. DNA unwinding element). Кроме того, существуют три дополнительных участка связывания DnaA (I -сайты) и участки связывания белков IHF (от англ. integration hose factor) и FIS (от англ. factor for inversion stimulation). Эти два белка необходимы для некоторых реакций рекомбинации, описанных ниже в данной главе, а их названия отражают их функции. Еще один ДНК-связывающий белок HU (гистоноподобный бактериальный белок, ранее называвшийся фактором U) также участвует в процессе, но не имеет специфического участка связывания.
Рис. 25-11. Расположение последовательностей в точке начала репликации Е. coli (oriC). Показаны консенсусные последовательности (см. с. 156 в т. 1) ключевых повторов. N обозначает любой из четырех нуклеотидов. Горизонтальные стрелки показывают ориентацию нуклеотидных последовательностей (стрелки слева направо указывают на последовательность в верхней цепи, а справа налево — в нижней). Участки FIS и IHF — сайты связывания белков, описанных в тексте. С R-сайтами связан белок DnaA. Сайты I — дополнительные участки связывания DnaA (у них другие последовательности). DnaA взаимодействует с ними только в комплексе с АТР.

В инициации репликации участвуют по меньшей мере 10 разных ферментов и белков (см. табл. 25-3). Они расплетают спираль ДHК в точке начала репликации и собирают препраймирующий комплекс для последующих реакций. Ключевым элементом в процессе инициации выступает белок DnaA — входит в белковое семейство ААА+ АТРаз (от англ. ATPases associated with diverse cellular activities). Многие AAA+ АТРазы, включая DnaA, образуют олигомерные структуры и сравнительно медленно гидролизуют АТР. Гидролиз АТР служит переключателем, опосредующим переходы одной формы белка в другую. В случае DnaA ATP-связанная форма активна, а ADP-связанная форма неактивна.
Восемь молекул белка DnaA, каждая из которых связана с АТР, образуют спиральный комплекс, охватывающий R- и I-сайты в oriC (рис. 25-12). DnaA имеет более высокое сродство к R-сайтам, чем к 1-сайтам, и этот белок связывается с R-сайтами одинаково хорошо как в АТР-, так и в ADP-форме. Напротив, 1-сайты, в которых связывается только ATP-связанный DnaA, позволяют различать активную и неактивную форму DnaA. Молекула ДНК плотно закручивается в правую сторону вокруг этого комплекса, образуя положительную суперспираль (см. гл. 24). Возникшее в соседних участках ДНК напряжение вызывает денатурацию А = Т-богатого участка DUE. Кроме того, образующийся в точке начала репликации комплекс также содержит несколько ДНК-связываюших белков — HU, IHF и FIS, которые облегчают изгиб ДНК.
Рис. 25-12. Модель инициации репликации в точке начала репликации Е. coli (оri’С). Восемь связанных с АТР молекул белка DnaA присоединяются к точке начала репликации в R- и I-сайтах (см. рис. 25-11). ДНК закручивается вокруг этого комплекса с образованием правозакрученной спирали. Богатый остатками А = Т участок DUE денатурирует из-за напряжения, возникающего в цепи в результате связывания DnaA. Образованию спирального комплекса с DnaA способствуют белки HU, IHF и FIS (здесь не показаны, так как подробно их структура и роль еще не определены). Гексамеры белка DnaB связываются с каждой цепью ДНК с помощью белка DnaC. Благодаря своей хеликазной активности белок DnaB раскручивает ДНК, подготавливая ее для посадки праймера и синтеза ДНК.

Затем другая ААА+ АТРаза, DnaC, загружает белок DnaB на разделенные цепи ДНК в денатурированном участке. Гексамер, состоящий из молекул DnaC, каждая из которых связана с АТР, образует прочный комплекс с гексамерной кольцевой DnaB-хеликазой. При взаимодействии DnaC с DnaB кольцо DnaB раскрывается, чему также способствует дальнейшее взаимодействие между DnaB и DnaA. Два кольцевых гексамера DnaB связываются с DUE — по одному на каждую нить ДНК. Связанная с DnaC молекула АТР гидролизуется, что приводит к высвобождению DnaC, a DnaB остается связанным с ДНК.
Присоединение хеликазы DnaB — ключевой момент в инициации репликации. Далее репликативная хеликаза DnaB перемещается вдоль одноцепочечной ДНК в направлении 5′ —> 3′, раскручивая ДНК по мере продвижения. Таким образом. связанные с двумя пенями ДНК молекулы хеликазы DnaB перемещаются в противоположных направлениях, создавая две репликативные вилки. Все другие белки репликативной вилки также напрямую или опосредованно связаны с DnaB. Холофермент ДНК-полимеразы III связан через Ʈ-субъединицы. Другие взаимодействия DnaB описаны ниже. В начале репликации, когда нити ДНК разделяются в области репликативной вилки, с ними связываются, стабилизируя их, многочисленные молекулы белка, связывающегося с одноцепочечной ДНК (single-stranded DNA-binding protein, SSB), а ДНК-гираза (ДНК- топоизомераза II) снимает топологическое напряжение, возникающее впереди вилки из-за раскручивания ДНК.
Инициация — единственная фаза репликации ДНК, про которую известно, что она регулируется, притом таким образом, что репликация происходит только один раз за клеточный цикл. Механизм регуляции пока еще точно не установлен. но в ходе генетических и биохимических исследований было выявлено несколько независимых элементов регуляции.
Как только с ДНК связывается ДНК-полимераза III, а также β-субъединицы (что свидетельствует о завершении фазы инициации), с β-субъединицами связывается белок Hda, который взаимодействует с DnaA и стимулирует гидролиз связанного с ним АТР. Белок Hda (название этого фермента означает «гомологичный DnaA») — еще одна ААА+ АТРаза, родственная DnaA. Гидролиз АТР приводит к разборке комплекса DnaA в точке начала репликации. Медленное высвобождение ADP и связывание новых молекул АТР замыкает цикл превращений между неактивной (со связанным ADP) и активной (со связанным АТР) формами белка, который длится от 20 до 40 мин.
На ход инициации репликации влияет метилирование ДНК и взаимодействия с бактериальной плазматической мембраной. ДНК в области оriС метилируется Dam-метилазой (от англ. DNA adenine methylation.; табл. 25-3), которая вводит метальную группу по положению N6 в остатке аденина в палиндромной последовательности (5’) GATC. Точка начала репликации оriС у Е. coli содержит особенно много повторов GATC — их 11 в последовательности из 245 п. н. тогда как в среднем во всей хромосоме Е. coli этот повтор встречается одни раз на 256 п. н.
Таблица 25-3. Белки, необходимые для инициации репликации в точке начала репликации E. coli
|
Белок |
Мr |
Число субъединиц |
Функция |
|
Белок DnaA |
52 000 |
1 |
Распознает последовательности в точке начала репликации; раскрывает цепи ДНК в специальных сайтах точки начала репликации |
|
Белок DnaB (хеликаза) |
300 000 |
6* |
Раскручивает ДНК |
|
Белок DnaC |
29 000 |
6* |
Связывает DnaB в точке начала репликации |
|
HU |
19 000 |
2 |
Гистоноподобный ДНК-связывающий белок; стимулирует инициацию |
|
FIS |
22 500 |
2* |
ДНК-связывающий белок; стимулирует инициацию |
|
IHF |
22 000 |
2 |
ДНК-связывающий белок; стимулирует инициацию |
|
Праймаза (белок DnaG) |
60 000 |
1 |
Синтезирует РНК-праймеры |
|
Белок, связывающий одноцепочечную ДНК (SSB) |
75 600 |
4* |
Связывает одноцепочечную ДНК |
|
ДНК-гираза (ДНК-топоизомераза II) |
400 000 |
4 |
Ослабляет торсионное напряжение, вызываемое раскручиванием ДНК |
|
Dam-метилаза |
32 000 |
1 |
Метилирует последовательность (5′)GATC в oriС |
* В данном случае субъединицы одинаковые.
Непосредственно после репликации ДНК оказывается наполовину метилированной; в родительских цепях последовательности оriС метилированы, а во вновь синтезированных — нет. Эти наполовину метилированные последовательности оriС изолированы за счет взаимодействия с плазматической мембраной (механизм неизвестен) и связывания с белком SeqA. Через некоторое время последовательность оriС отделяется от плазматической мембраны, SeqA диссоциирует, и Dam-метилаза полностью метилирует ДНК до того, как она вновь связывает DnaA в новом раунде репликации.
Элонгация.
Фаза элонгации репликации включает две связанные между собой операции: синтез лидирующей цепи и синтез отстающей цепи. Для синтеза обеих цепей важно присутствие в репликатвной вилке нескольких ферментов. Сначала исходная ДНК раскручивается ДНК-хеликазой, а возникающее топологическое напряжение снимается топоизомеразами. Затем белок SSB стабилизирует каждую отдельную цепь. С этого момента синтез лидирующей цени и синтез отстающей цепи кардинально различаются.
Синтез лидирующей цепи происходит проще: он начинается с синтеза праймазой (белок DnaG) короткого (от 10 до 60 нуклеотидов) РНК- праймера в точке начала репликации. Для осуществления данной реакции DnaG взаимодействует с хеликазой DnaB. причем праймер синтезируется в направлении, которое противоположно направлению движения хеликазы DnaB. На самом деле хеликаза DnaB перемещается вдоль цепи, которая становится отстающей при синтезе ДНК. Однако первый праймер, синтезированный при первом взаимодействии DnaG-DnaB, служит для запуска синтеза лидирующей цепи ДНК в противоположном направлении. Дезоксирибонуклеотиды добавляются к этому праймеру ДНК-полимеразой III, связанной с DnaB-хеликазой, расположенной на противоположной цепи ДНК. Синтез лидирующей цепи происходит непрерывно со скоростью, соответствующей скорости раскручивания ДНК в репликативной вилке.
Как уже было отмечено, отстающая цепь синтезируется в виде коротких фрагментов Оказаки (рис. 25-13, а). Сначала праймаза синтезирует РНК-праймер. Затем, как и в процессе синтеза лидирующей цепи, ДНК-полимераза III связывается с праймером и присоединяет дезоксирибонуклеотиды (рис. 25-13, б). На этом этапе синтез каждого фрагмента Оказаки кажется простым, но на самом деле это весьма сложный процесс. Сложность заключается в координации синтезов лидирующей и отстающей цепей: обе цепи синтезируются одним асимметричным димером ДНК-полимеразы III, что достигается при образовании петли ДНК из отстающей цепи (рис. 25-14), сближающей две точки полимеризации.
Рис. 25-13. Синтез фрагментов Оказаки, а — на определенном расстоянии праймаза синтезирует РНК-праймер для нового фрагмента Оказаки. Если рассматривать две параллельные матричные цепи, синтез отстающей цепи формально происходит в направлении, противоположном направлению движения вилки, б — каждый праймер удлиняется ДНК-полимеразой III. в — синтез ДНК продолжается до тех пор, пока полимераза не достигнет праймера предыдущего фрагмента Оказаки. Для возобновления процесса вблизи репликативной вилки синтезируется новый праймер.

Синтез фрагментов Оказаки на отстающей цепи происходит с участием удивительного ферментативного аппарата. DnaB-хеликаза и DnaG- праймаза образуют отдельный функциональный блок, праймосому, в составе репликативного комплекса. ДНК-полимераза III использует один набор кор-ферментов (коровая полимераза) для непрерывного синтеза лидирующей цепи, в то время как другой набор кор-ферментов осуществляет циклы синтеза фрагментов Оказаки на петле отстающей цепи. DnaB-хеликаза, находящаяся перед ДНК-полимеразой III, раскручивает ДНК в репликативной вилке (рис. 25-14, а), продвигаясь вдоль матрицы отстающей цепи в направлении 5′ —> 3′. ДНК-праймаза время от времени связывается с DnaB-хеликазой и синтезирует короткий РНК-праймер (рис. 25-14, б). Затем погрузчик зажима ДНК-полимеразы III закрепляет на праймере новый скользящий β-зажим (рис. 25-14, в). Когда синтез фрагмента Оказаки заканчивается, репликация приостанавливается, кор-фермент ДНК-полимеразы III отсоединяется от одного зажима (а также от законченного фрагмента Оказаки) и связывается с новым зажимом (рис. 25-14, г, б). Это инициирует синтез нового фрагмента Оказаки. Как отмечалось ранее, весь комплекс, ответственный за координированный синтез ДНК в репликатиной вилке, называют реплисомой. Белки, задействованные в репликативной вилке, перечислены в табл. 25-4.
Таблица 25-4. Белки реплисомы E. coli
|
Белок |
Мr |
Число субъединиц |
Функция |
|
SSB |
75 600 |
4 |
Связывание с одноцепочечной ДНК |
|
Белок DnaB (хеликаза) |
300 000 |
6 |
Раскручивание ДНК; компонент праймосомы |
|
Праймаза (белок DnaG) |
60 000 |
1 |
Синтез РНК-праймера; компонент праймосомы |
|
ДНК-полимераза III |
791 500 |
17 |
Элонгация новой цепи |
|
ДНК-полимераза I |
103 000 |
1 |
Заполнение брешей; вырезание праймеров |
|
ДНК-лигаза |
74 000 |
1 |
Лигирование |
|
ДНК-гираза (ДНК-топоизомераза II) |
400 000 |
4 |
Суперскручивание |
Рис. 25-14. Синтез ДНК на лидирующей и отстающей цепях. События, происходящие в репликативной вилке, координируются одним димером ДНК-полимеразы III в комплексе с DnaB-хеликазой. Здесь показан уже идущий процесс репликации (стадии от а до д обсуждаются в тексте). Отстающая цепь изогнута петлей, так что синтез ДНК происходит одновременно на лидирующей и на отстающей матрице. Красные стрелки указывают 3′-концы двух новых цепей и направление синтеза ДНК, толстые черные стрелки — направление движения родительской ДНК через комплекс. На отстающей цепи начинает синтезироваться фрагмент Оказаки. Цветовое обозначение субъединиц и функции погрузчика зажима объясняются на рис. 25-15.

Погрузчик зажима ДНК-полимеразы III. состоящий из частей двух Ʈ-субъединиц, а также из y, δ и δ’-субъединиц, функционирует, кроме того, как ААА+ АТРаза. Этот комплекс связывается с АТР и с новым скользящим β-зажимом. Связывание приводит к возникновению напряжения в димерном зажиме, в результате чего на границе одной из субъединиц открывается кольцо (рис. 25-15). Отстающая цепь с только что присоединенным праймером проскальзывает в кольцо через образующийся разрыв. Затем погрузчик зажима гидролизует АТР, высвобождая скользящий (3-зажим и позволяя ему сомкнуться вокруг ДНК.
Рис. 25-15. Комплекс погрузчика зажима ДНК-полимеразы III. Комплекс образован пятью субъединицами: субъединица y, δ и δ’, а также N-концевыми доменами обеих Ʈ-субъединиц (см. рис. 25-10). Комплекс связывает три молекулы АТР и димерный β-зажим. Это связывание приводит к открыванию β-зажима на границе одной из двух субъединиц. Гидролиз связанного АТР позволяет β-зажиму вновь замкнуться вокруг ДНК.

Реплисома осуществляет синтез ДНК быстро, присоединяя примерно по 1000 нуклеотидов в секунду к каждой цепи (лидирующей и отстающей). После завершения сборки одного фрагмента Оказаки его РНК-праймер удаляется и замещается последовательностью ДНК с помощью ДНК-полимеразы I, а оставшийся ник ликвидируется ДНК-лигазой (рис. 25-16).
Рис. 25-16. Заключительные стадии синтеза фрагментов отстающей цепи. ДНК-полимераза I благодаря своей 5′ —> 3′-экзонуклеазной активности удаляет РНК- праймеры из отстающей цепи и замещает их на ДНК. Оставшийся ник (разрыв) ликвидирует ДНК-лигаза. Роль АТР и NAD+ показана на рис. 25-17.

ДНК-лигаза катализирует образование фосфодиэфирной связи между 3’-гидроксильной группой на конце одной цепи ДНК и 5’-фосфатом на конце другой цепи. Фосфат должен быть предварительно активирован путем аденилирования. ДНК-лигазы, выделенные из вирусов и эукариот, используют для этой цели АТР. ДНК-лигазы бактерий (рис. 25-17) отличаются тем, что в качестве источника АМР они используют NAD+- кофактор, который обычно участвует в реакциях с переносом гидрид-иона (см. рис. 13-24). ДНК- лигаза еще один фермент метаболизма ДНК, который стал важным инструментом в экспериментах с рекомбинантными ДНК (см. рис. 9-1).
Рис. 25-17. Механизм реакции. Действие ДНК-лигазы. На каждой из трех стадий одна фосфодиэфирная связь образуется за счет другой. Стадии ① и ② приводят к активации 5′-фосфатной группы в нике. АМР сначала переносится на остаток лизина в молекуле фермента, а затем на 5′-фосфат в нике. На стадии ③ 3′-гидроксильная группа атакует этот фосфат и вытесняет АМР, образуя фосфодиэфирную связь и закрывая тем самым разрыв. У Е. coli АМР в реакции ДНК-лигазы происходит из NAD+. ДНК- лигазы, выделенные из некоторых вирусных и эукариотических источников, используют АТР, а не NAD+, и на стадии ① они высвобождают пирофосфат, а не никотина- мидмононуклеотид (NMN).

Терминация.
Наконец, две репликативные вилки кольцевой хромосомы Е. coli встречаются в точке терминации, содержащей множество копий последовательности из 20 п. н« называемой Теr (от лат. terminus — конец) (рис. 25-18). Последовательности Теr расположены на хромосоме таким образом, что создают что-то вроде ловушки, в которую репликативная вилка может войти, но не может из нее выйти. Теr-последовательности служат участками связывания белка Tus (от англ. terminus utilization substance). Комплекс Tus Ter может задержать репликативную вилку, движущуюся только в одном направлении. В каждом цикле репликации действует только один комплекс Tus- Теr — первый из тех, с которым столкнется первая вилка. Учитывая, что движущиеся в противоположных направлениях репликативные вилки обычно останавливаются при встрече, может показаться, что Теr-последовательности — необязательные элементы. Однако они могут предотвратить избыточную репликацию на одной из репликативных вилок, в случае если другая задерживается или останавливается при столкновении с повреждением ДНК или каким-либо другим препятствием.
Рис. 25-18. Терминация репликации хромосомы Е. coli. Последовательности Теr (от ТеrА до TerF) располагаются на хромосоме в виде двух кластеров с противоположной ориентацией.

Таким образом, когда одна из репликативных вилок встречает функциональный комплекс Tus-Ter, она останавливается; а другая вилка останавливается, когда встречается с первой (задержанной) вилкой. Затем реплицируются последние несколько сотен пар оснований ДНК между этими крупными белковыми комплексами (по неизвестному механизму), достраивая две топологически сцепленные (катенированные) кольцевые хромосомы (рис. 25-19). Кольцевые ДНК, связанные таким образом, называются катенанами. Функцию разделения катенированных колец у Е. coli выполняет топоизомераза IV (топоизомераза II типа). Затем при клеточном делении разделенные хромосомы расходятся по дочерним клеткам. Заключительная фаза репликации других кольцевых хромосом, включая многие из ДНК-содержащих вирусов, инфицирующих эукариотические клетки, происходит по аналогичной схеме.
Рис. 25-19. Роль топоизомераз в терминации репликации. В результате репликации ДНК на двух противоположно направленных репликативных вилках образуются полноценные хромосомы, соединенные как катанены (топологически соединенные кольца). Кольца не связаны ковалентной связью, но поскольку они переплетены и каждое из них замкнуто ковалентно, их нельзя разделить без помощи топоизомераз. У Е. coli основную роль в разделении катенированных хромосом играет топоизомераза типа II (ДНК-топоизомераза IV), которая вносит кратковременный разрыв в обе цепи ДНК в одной хромосоме, позволяя другой хромосоме пройти сквозь разрыв.

Репликация в эукариотических клетках происходит по похожей схеме, но сложнее
Молекулы ДНК в эукариотических клетках значительно крупнее, чем в бактериях, и образуют сложные нуклеопротеиновые структуры (хроматин; разд. 24.3). Основная схема репликации ДНК у эукариот такая же, как у бактерий, и многие белковые комплексы похожи по функциям и структуре. Однако репликация у эукариот регулируется и координируется в соответствии с клеточным циклом, что усложняет процесс.
Участки начала репликации достаточно хорошо охарактеризованы у некоторых низших эукариот, но значительно хуже у высших эукариот. У позвоночных для инициации репликации могут использоваться разные А = Т-богатые последовательности, причем участки начала репликации могут меняться при каждом клеточном делении. Репликация дрожжей Saccharomyces cerevisiae начинается в определенной области, называемой автономно реплицирующейся последовательностью (АРП) или репликатором. Дрожжевые репликаторы имеют длину примерно 150 п. н. и содержат несколько важных консервативных последовательностей. В 16 хромосомах гаплоидного генома дрожжей рассредоточено около 400 репликаторов.
Механизмы регуляции обеспечивают однократную репликацию всей клеточной ДНК в каждом клеточном цикле. Важную роль в этой регуляции играют белки, называемые циклинами, и циклин-зависимые киназы (CDK), с которыми они образуют комплексы (с. 660 в т. 1). Циклины быстро разрушаются в результате убиквитин- зависимого протеолиза в конце М-фазы (фазы митоза), а отсутствие циклинов способствует организации пререпликативных комплексов (рrе- RC) в участках инициации репликации. В быстро растущих клетках комплекс pre-RC образуется в конце М-фазы. В медленно растущих клетках этот комплекс не образуется до конца фазы G1. Формирование комплексов pre-RC позволяет клетке начать репликацию (этот этап иногда называют лицензированием).
Как и у бактерий, ключевой момент в инициации репликации у всех эукариот — присоединение репликативной хеликазы, гетерогексамерного белкового комплекса МСМ (от англ. minichromosome maintenance; субъединицы от МСМ2 до МСМ7). Кольцевая хеликаза, состоящая из субъединиц МСМ2-7, функционирует подобно бактериальной хеликазе DnaB и связывается с ДНК при помощи другого комплекса из шести белков, называемого комплексом распознавания точки инициации репликации (ORC; от англ. origin recognition complex; рис. 25-20). Комплекс ORC состоит из пяти ААА+ АТРазных доменов и по своим функциям напоминает бактериальный комплекс DnaA. Кроме того, для присоединения комплекса МСМ2-7 необходимы два белка — CDC6 (от англ. cell division cycle) и CDT1 (от англ. CDC 10-dependent transcript 1), причем дрожжевой белок CDC6 представляет собой еще одну ААА+ АТРазу.
Рис. 25-20. Сборка пререпликативного комплекса в точке начала репликации у эукариот. В участке инициации репликации связываются белки ORC, CDC6 и CDT1. Эти белки, многие из которых являются ААА+ АТРазами, способствуют присоединению репликационной хеликазы МСМ2-7 в реакции, аналогичной реакции присоединения бактериальной хеликазы DnaB с помощью белка DnaC. В результате связывания хеликазного комплекса МСМ с ДНК образуется пререпликативный комплекс pre-RC, и именно эта стадия оказывается ключевой для инициации репликации.

Для осуществления репликации в S-фазе происходит синтез и активация комплексов циклин-CDK (таких, как комплекс циклин Е- CDK2; см. рис. 12-45 в т. 1) и CDC7-DBF4. Оба типа комплексов помогают активизировать репликацию путем связывания и фосфорилирования некоторых белков пререпликативных комплексов. Другие циклины и CDK ингибируют образование дополнительных pre-RC комплексов после начала репликации. Например, CDK2 связывается с циклином А, когда уровень циклина Е снижается в ходе S-фазы, что ингибирует активность CDK2 и предотвращает образование дополнительных pre-RC-комплексов.
Скорость репликативной вилки у эукариот (около 50 нуклеотидов в секунду) примерно в 20 раз меньше, чем в Е. coli. Если бы репликация хромосомы человека начиналась в единственной точке инициации репликации, при такой скорости процесса репликация средней хромосомы продолжалась бы более 500 ч. На самом деле репликация хромосом человека происходит в двух направлениях и начинается во многих точках, удаленных друг от друга на расстояние от 30 до 300 т. п. н. Хромосомы эукариот почти всегда намного длиннее, чем хромосомы бактерий, поэтому наличие многих точек инициации репликации, вероятно, общее свойство клеток эукариот.
Как и у бактерий, у эукариот есть несколько типов ДНК-полимераз. Некоторые из них, возможно, выполняют особые функции, например, репликацию митохондриальной ДНК. В репликации ядерных хромосом участвует ДНК-полимераза а в комплексе с ДНК-полимеразой δ. Субъединичная ДНК-полимераза α имеет похожую структуру и свойства во всех эукариотических клетках. Одна из ее субъединиц выполняет функцию праймазы, а самая крупная субъединица (Мr ≈ 180 000) обладает полимеразной активностью. Однако данная полимераза не имеет корректирующей 3′ —> 5′-экзонуклеазной активности и не может обеспечить высокой точности репликации. Предполагается, что ДНК-полимераза α нужна только для синтеза коротких праймеров (либо РНК, либо ДНК) для фрагментов Оказаки на отстающей цепи. Удлинение этих праймеров осуществляет мультисубъединичная ДНК -полимераза δ. Этот фермент связывается с белком, называемым ядерным антигеном пролиферирующих клеток (PCNA — от англ. proliferating cell nuclear antigen; Mr = 29 000), и стимулируется им; этот белок в большом количестве содержится в ядрах делящихся клеток. Трехмерная структура PCNA удивительным образом напоминает структуру β-субъединицы ДНК-полимеразы III Е. соli (рис. 25-10, б), хотя первичные последовательности этих белков не имеют выраженной гомологии. Функционально PCNA представляет собой аналог β-субъединиц; он формирует кольцевой зажим, который существенно увеличивает процессивность полимеразы. ДНК-полимераза δ обладает корректирующей 3′ —> 5′-экзонуклеазной активностью и, предположительно, обеспечивает синтез как лидирующей, так и отстающей цепи в комплексе, напоминающем димерную бактериальную ДНК-полимеразу III.
Еще одна полимераза, ДНК-полимераза ε, в некоторых ситуациях заменяет ДНК-полимеразу δ, например, при репарации ДНК. ДНК-полимераза е также может действовать в репликативной вилке, возможно, аналогично бактериальной ДНК-полимеразе I, удаляя праймеры фрагментов Оказаки на отстающей цепи.
В репликации ДНК у эукариот задействованы еще два белковых комплекса. Белок RPA (от англ. replication protein А) связывает одноцепочечную ДНК, и его функция аналогична функции белка SSB в клетках Е. coli Белок RFC (от англ/ replication factor С) представляет собой погрузчик зажима для PCNA, который облегчает сборку активных репликационных комплексов. Аминокислотные последовательности субъединиц комплекса RFC имеют значительное сходство с последовательностями субъединиц бактериального y-комплекса.
Для терминации репликации линейных эукариотических хромосом на концах каждой хромосомы синтезируются специальные структуры, называемые теломерами (см. гл. 26).
Вирусные ДНК-полимеразы являются мишенями для противовирусной терапии
Многие ДНК-co держащие вирусы кодируют собственные ДНК-полимеразы, и некоторые из них становятся мишенями для лекарственных средств. Например, ДНК-полимераза вируса простого герпеса ингибируется ацикловиром, разработанным Гертрудой Элайон (см. с. 558 в т. 2). Ацикловир состоит из гуанина, к которому присоединено неполное кольцо рибозы.

Ацикловир фосфорилируется вирусной тимидинкиназой. Сродство ацикловира к вирусному ферменту в 200 раз выше сродства к клеточной тимидинкиназе. В результате фосфорилирование происходит преимущественно в клетках, инфицированных вирусом. Клеточные киназы превращают образовавшийся ацикло-GMP в ацикло-GTP, который одновременно является и ингибитором, и субстратом ДНК-полимераз. Ацикло-GTP сильнее ингибирует ДНК-полимеразу вируса герпеса, чем клеточные ДНК-полимеразы. Поскольку ацикло-GTP не имеет 3′-гидроксильной группы, его включение в цепь ДНК воспринимается как сигнал терминации. Таким образом, репликация вируса ингибируется на нескольких стадиях. ■
Краткое содержание раздела 25.1 Репликация ДНК
■ Репликация ДНК происходит с очень высокой точностью и в определенной фазе клеточного цикла. Репликация полуконсервативна, каждая цепь выступает матрицей для новой дочерней цепи. Репликация протекает в три стадии: инициация, элонгация и терминация. В бактериях реакция начинается в точке инициации репликации и обычно происходит в двух направлениях.
■ ДНК синтезируется ДНК-полимеразами в направлении 5′ —> 3′. В репликативной вилке лидирующая цепь синтезируется непрерывно по ходу движения репликативной вилки; отстающая цепь синтезируется прерывисто в виде фрагментов Оказаки, которые затем сшиваются.
■ Точность репликации ДНК достигается (1) путем выбора оснований полимеразой, (2) с помощью корректирующей 3′ —> 5′-экзонуклеазной активности, которой обладает большинство ДНК-полимераз, и (3) с помощью специфических систем репарации, исправляющими ошибки, оставшиеся после репликации.
■ Большинство клеток имеет несколько ДНК-полимераз. У Е. coli основной фермент репликации — ДНК-полимераза III. ДНК-полимераза I выполняет особые функции в ходе репликации, рекомбинации и репарации.
■ На стадиях инициации, элонгации и терминации репликации ДНК работает множество ферментов и белковых факторов, многие из них принадлежат к семейству ААА+ АТРаз.
■ Белки бактерий, участвующие в репликации, организованы в крупные комплексы, внутри которых матрица ДНК протягивается через две реплисомы, связанные с плазматической мембраной.
ДНК-полимераза — фермент,
участвующий в репликации
ДНК.
Ферменты этого класса катализируют
полимеризацию дезоксирибонуклеотидов вдоль
цепочки нуклеотидов ДНК,
которую фермент «читает» и использует
в качестве шаблона. Тип нового нуклеотида
определяется по принципу
комплементарности с
шаблоном, с которого ведётся считывание.
Собираемая молекула комплементарна
шаблонной моноспирали и идентична
второму компоненту двойной спирали.
Выделяют ДНК-зависимую
ДНК-полимеразу (КФ 2.7.7.7),
использующую в качестве матрицы одну
из цепей ДНК, и РНК-зависимую
ДНК-полимеразу (другое
название обратная
транскриптаза, КФ 2.7.7.49),
способную также к считыванию информации
с РНК (обратная
транскрипция).
ДНК-полимеразу
считают холоферментом,
поскольку для нормального функционирования
она требует присутствия ионов магнияв
качестве кофактора. В отсутствие ионов
магния о ней можно говорить как
об апоферментe.
ДНК-полимераза
начинает репликацию ДНК, связываясь с
отрезком цепи нуклеотидов. Среднее
количество нуклеотидов, присоединяемое
ферментов ДНК-полимеразой за один акт
связывания/диссоциации с матрицей,
называют процессивностью.
Действие
ДНК-полимеразы
Репликация
ДНК
Как
известно, две цепи молекулы ДНК
антипараллельны. Разные концы одной
цепи называются 3’-конец и 5’-конец.
Репликация происходит путем непрерывного
роста нуклеотида за нуклеотидом обеих
новых цепей одновременно. Матрица
считывается ДНК-полимеразой только в
направлении 3’-5’, добавляя свободные
нуклеотиды к 3’-концу собираемой цепочки.
Поэтому синтез ДНК происходит непрерывно
только на одной из матричных цепей,
называемой «лидирующей».
Во второй цепи («отстающей»)
синтез происходит короткимифрагментами.
Ни
одна из известных ДНК-полимераз не может
создать цепочку «с нуля»: они в состоянии
лишь добавлять нуклеотиды к уже
существующей 3’-гидроксильной группе.
По этой причине ДНК-полимераза нуждается
впраймере,
к которому она могла бы добавить первый
нуклеотид. Праймеры состоят из
оснований РНК и
ДНК, при этом первые два основания всегда
РНК-основания. Праймеры синтезируются
другим ферментом — праймазой.
Ещё один фермент — геликаза —
необходим для раскручивания двойной
спирали ДНК с формированием одноцепочечной
структуры, которая обеспечивает
репликацию обеих цепочек в соответствии
с полуконсервативной моделью репликации
ДНК.
Некоторые
ДНК-полимеразы обладают также способностью
исправлять ошибки во вновь собираемой
цепочке ДНК. Если происходит обнаружение
неправильной пары нуклеотидов,
ДНК-полимераза откатывается на один
шаг назад. Благодаря своей
3′-5′ экзонуклеазной гидролитической
активности ДНК-полимераза может исключить
неправильный нуклеотид из цепочки и
затем вставить на его место правильный,
после чего репликация продолжается в
нормальном режиме.
Многообразие
ДНК-полимераз
Структура
ДНК-полимераз достаточно жёстко
фиксирована. Их каталитические субъединицы
очень мало различаются в различных
видах живых клеток. Такая фиксация
структуры обычно появляется там, где
отсутствие разнообразия обусловлено
огромной важностью или даже незаменимостью
для функционирования клетки.
Генами
некоторых вирусов тоже
кодируются особые ДНК-полимеразы,
которые могут избирательно реплицировать
вирусные ДНК. Ретровирусы обладают
геном необычной ДНК-полимеразы, называемой
ещё обратной
транскриптазой,
являющейся РНК-зависимой ДНК-полимеразой
и осуществляющей сборку ДНК на основе
шаблонной РНК.
Семейства
ДНК-полимераз
На
основании своей структуры ДНК-полимеразы
могут быть разбиты на семь различных
семейств: A, B, C, D, X, Y, и RT.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Механизмы исправления ошибок во время репликации ДНК и ее репарация вследствие повреждений на протяжении всего жизненного цикла клетки.
Основные моменты:
-
Клетки имеют различные механизмы предотвращения возникновения мутаций – необратимых изменений в ДНК
-
В процессе синтеза ДНК, большинство ДНК-полимераз «проверяют свою работу» и проводят замену бо́льшей части ошибочно вставленных нуклеотидов. Этот процесс можно назвать исправлением ошибок.
-
Сразу после синтеза ДНК любые оставшиеся ошибочные нуклеотиды обнаруживаются и заменяются в так называемом процессе репарации ошибочно спаренных нуклеотидов.
-
Если ДНК повреждена, она может быть восстановлена с помощью различных механизмов, например, путём прямой репарации, эксцизионной репарации или путём восстановления двухцепочечных разрывов
- пострепликативной репарации.
Введение
Как ДНК связана с раком? Рак возникает при неконтролируемом делении клеток, когда игнорируются клеточные «стоп»-сигналы, что приводит к образованию опухоли. Это неправильное поведение клеток вызвано накопившимися мутациями — необратимыми изменениями последовательности ДНК клетки.
На самом деле, ошибки в процессе репликации и повреждения ДНК возникают в клетках нашего тела постоянно. Однако в большинстве случаев они не приводят к раку и даже не вызывают мутаций, такие ошибки обычно обнаруживаются и исправляются в процессе репарации ДНК. Если же повреждение исправить не удаётся, то в клетке включается механизм самоуничтожения — (апоптоз), который предотвращает передачу поврежденной ДНК дочерним клеткам.
Мутации возникают и передаются дочерним клеткам только тогда, когда эти механизмы не справляются. В частности, рак возникает в случае накопившихся в одной клетке мутаций генов, связанных с делением.
В этой статье мы подробно рассмотрим механизмы, используемые клетками для исправления ошибок, которые возникают в процессе репликации. К ним относятся:
-
Исправление ошибок – процесс, который возникает во время репликации ДНК.
-
Репарация ошибочно спаренных нуклеотидов, которая происходит сразу же после репликации ДНК.
-
Механизмы репарации, которые выявляют и исправляют повреждения ДНК на протяжении всего клеточного цикла
Исправление ошибок
ДНК-полимеразы — это ферменты, участвующие в репликации ДНК. Во время копирования ДНК большинство ДНК-полимераз «проверяют», корректный ли нуклеотид они добавляют. Этот процесс называется исправлением ошибок. Если полимераза обнаружит, что был добавлен неправильный нуклеотид, она сразу же удалит и заменит его и только после этого продолжит синтез ДНКstart superscript, 1, end superscript.
Репарация ошибочно спаренных нуклеотидов
Процесс исправления избавляет от основной массы ошибок, но не от всех. После создания новой ДНК запускается механизм репарации ошибочно спаренных нуклеотидов — удаления и замены ошибочно спаренных нуклеотидов, оставшихся в результате репликации. Исправление несоответствий между парами оснований также может включать в себя исправление небольших вставок и делеций, возникающих вследствие «соскальзывания» полимеразы с исходной цепи squared.
Как происходит восстановление неправильно спаренных нуклеотидов? Во-первых, белковый комплекс распознаёт неправильно спаренный нуклеотид и связывается с ним. Другой комплекс разрезает ДНК в области несовпадения, а ещё одна группа ферментов отщепляет некорректный нуклеотид вместе с небольшим участком вокруг него. Затем ДНК-полимераза заполняет этот пробел правильными нуклеотидами, а фермент ДНК-лигаза сшивает разрывы в цепиsquared.
Удивительно: как белки, участвующие в восстановлении ДНК, определяют, «кто прав» во время репарации ошибочно спаренных нуклеотидов? То есть, когда два основания неправильно соединены (как G (гуанин) и T (тимин) на рисунке выше), какое из этих двух оснований должно быть удалено и заменено?
У бактерий можно отличить исходную и дочернюю цепи ДНК по метилированным основаниям. На исходной цепи ДНК есть метильные (minus, start text, C, H, end text, start subscript, 3, end subscript) группы, присоединенные к некоторым из ее оснований, а у дочерней цепи таких групп еще нетcubed.
У эукариот процессы, позволяющие идентифицировать исходную цепь при устранении несоответствий, включают распознавание одноцепочечных разрывов, которые обнаруживаются только у дочерней цепи cubed.
Механизмы репарации ДНК
С ДНК может что-нибудь случиться практически в любой момент жизни клетки, а не только во время репликации. Фактически, ДНК постоянно повреждается из-за воздействия внешних факторов: ультрафиолетового излучения и радиации, химических веществ, не говоря уже о спонтанных процессах, которые протекают даже без вмешательства окружающей среды!start superscript, 4, end superscript
К счастью, наши клетки имеют механизмы восстановления, с помощью которых они находят и исправляют большинство повреждений ДНК. Можно выделить несколько типов репарации:
-
Прямая репарация. Некоторые повреждения ДНК, вызванные химическими реакциями, могут быть «исправлены» находящимися в клетке ферментами.
-
Эксцизионная репарация. Повреждение одного или нескольких нуклеотидов ДНК часто исправляется удалением и заменой поврежденного участка. При эксцизионной репарации оснований удаляется только поврежденное основание. В случае эксцизионной репарации нуклеотидов, как и в случае репарации ошибочно спаренных нуклеотидов, которое мы рассмотрели выше, удаляются целиком нуклеотиды.
-
Репарация двухцепочечных разрывов: Существуют два основных способа: негомологичное соединение концов и гомологичная рекомбинация. Они используются для восстановления двухцепочечных разрывов ДНК (когда вся хромосома разделяется на две части).
Прямая репарация
В некоторых случаях клетка может исправить повреждение ДНК, обратив вызвавшую его реакцию. Дело в том, что «повреждение ДНК» — это, как правило, присоединение к ней лишней группы в результате химической реакции.
Например, гуанин (G) может подвергаться реакции с присоединением метильной (minus, start text, C, H, end text, start subscript, 3, end subscript) группы к атому кислорода в азотистом основании. Если это не исправить, метил-содержащий гуанин будет связываться с тимином (Т), а не с цитозином (С) во время репликации ДНК. К счастью, у людей и многих других организмов есть фермент, который может удалить метильную группу, обратив реакцию, и тем самым вернуть азотистое основание в нормальное состояниеstart superscript, 5, end superscript.
Эксцизионная репарация оснований
Эксцизионная репарация оснований — это механизм, используемый для обнаружения и удаления определенных типов поврежденных азотистых оснований. Ключевую роль в нем играет группа ферментов, называемых гликозилазами. Каждая гликозилаза обнаруживает и удаляет определенный вид поврежденных оснований.
Например, в процессе реакции дезаминирования цитозин может превратиться в урацил — основание, обычно встречающееся только в РНК. Во время репликации ДНК урацил будет соединяться с аденином, а не с гуанином (в отличие от цитозина), поэтому такое превращение может привести к возникновению мутацииstart superscript, 5, end superscript.
Для предотвращения подобных изменений гликозилаза, являющаяся частью сигнального пути эксцизионной репарации, обнаруживает и удаляет дезаминированные цитозины. После того, как основание было удалено, удаляется и оставшаяся часть нуклеотида, а другие ферменты заполняют пробелstart superscript, 6, end superscript.
Эксцизионная репарация нуклеотидов
Эксцизионная репарация нуклеотидов — это еще один способ удаления и замены поврежденных оснований. В результате нее обнаруживаются и корректируются повреждения, которые искажают форму двойной спирали ДНК. Например, азотистые основания могут измениться, присоединив к себе громоздкие группы атомов, в частности, в результате воздействия химических веществ, содержащихся в сигаретном дымеstart superscript, 7, end superscript.
Эксцизионная репарация нуклеотидов также используется для устранения повреждений, вызванных ультрафиолетовым излучением, например, при получении солнечного ожога. Под воздействием УФ-излучения цитозин и тимин могут вступать в реакцию с соседними основаниями, которые также являются цитозином или тимином, образуя при этом связи, изменяющие форму двойной спирали и вызывающие ошибки в процессе репликации ДНК. Наиболее распространенный тип таких связей — тиминовый димер — он состоит из двух тиминовых оснований, вступающих в реакцию друг с другом и образующих химическую связьstart superscript, 8, end superscript.
При эксцизионной репарации нуклеотидов поврежденные нуклеотиды удаляются вместе с соседними нуклеотидами. В этом процессе хеликаза (фермент, раскручивающий ДНК) раскрывает ДНК, образуя пузырь, а ферменты, разрезающие ДНК, отсекают поврежденную часть пузыря. Полимераза заполняет пробел, а лигаза сшивает разрыв в цепиstart superscript, 9, end superscript.
Репарация двухцепочечных разрывов
Некоторые факторы окружающей среды, например, радиация, могут вызывать разрывы обеих цепочек ДНК (разделение хромосомы на две части). Такие повреждения ДНК, если верить комиксам, ведут к появлению супергероев, но могут встречаться и после реальных катастроф, например, Чернобыльской.
Двухцепочечные разрывы опасны, потому что большие сегменты хромосом и сотни содержащихся в них генов могут быть потеряны, если разрыв не будет восстановлен. Существует два способа восстановления двухцепочечных разрывов ДНК: негомологичное соединение концов и гомологичная рекомбинация.
При негомологичном соединении концов два разорванных конца хромосомы просто склеиваются обратно. Этот механизм восстановления является «грубым» и неточным, в результате в месте разрыва, как правило, либо теряются нуклеотиды, либо добавляются лишние, что может привести к мутациям. Но это в любом случае лучше потери целого фрагмента хромосомыstart superscript, 10, end superscript.
При гомологичной рекомбинации для восстановления разрыва используется фрагмент из гомологичной хромосомы, который соответствует поврежденной хромосоме (или из сестринской хроматиды, если ДНК была реплицирована). В этом процессе две хромосомы объединяются, и неповрежденная область гомологичной хромосомы или хроматиды используется в качестве матрицы для замены поврежденной области. Гомологичная рекомбинация работает «чище», точнее, чем негомологичное соединение концов, и обычно не приводит к образованию мутацийstart superscript, 11, end superscript.
Репарация ДНК и заболевания человека
Доказательства важности механизмов репарации получены на основе генетических заболеваний человека. Во многих случаях мутации в генах, которые кодируют белки, участвующие в репарации, связаны с наследственным раком. Например:
-
Наследственный неполипозный колоректальный рак (также называемый синдромом Линча) вызван мутациями в генах, кодирующих белки, которые участвуют в репарации ошибочно спаренных нуклеотидовstart superscript, 12, comma, 13, end superscript. Поскольку такие нуклеотиды не восстанавливаются, у людей, страдающих этим синдромом, мутации накапливаются гораздо быстрее, чем у здоровых. Это может привести к развитию опухолей толстой кишки.
-
Люди с пигментной ксеродермой очень чувствительны к ультрафиолетовому излучению. Это вызвано мутациями в белках, участвующих в эксцизионной репарации нуклеотидов. Когда они не функционируют, димеры тимина и другие виды повреждений, вызванные ультрафиолетовым излучением, перестают восстанавливаться. У людей с пигментной ксеродермой после нескольких минут пребывания на солнце могут возникнуть сильные солнечные ожоги, и около половины из них заболевают раком кожи в возрасте до 10 лет, если только они не избегают солнечных лучейstart superscript, 14, end superscript.
ЧАСТЬ III. ПУТИ ПЕРЕДАЧИ ИНФОРМАЦИИ
Скучных ферментов не бывает.
— Артур Корнберг, «С любовью к ферментам», 1975
25. МЕТАБОЛИЗМ ДНК
В качестве хранилища генетической информации ДНК занимает центральное место среди биологических макромолекул. Нуклеотидная последовательность ДНК кодирует первичную структуру всех молекул клеточной РНК и белков и через ферменты опосредованно влияет на синтез всех других клеточных компонентов. Переход информации от ДНК к РНК и белку определяет размер, форму и жизнедеятельность всех живых существ.
ДНК прекрасно обеспечивает стабильное хранение генетической информации. Однако понятие «стабильное хранение» означает статичную неизменную картину. Оно не отражает сложности процессов, с помощью которых генетическая информация сохраняется и передается от одного поколения клеток к следующему. Метаболизм ДНК включает как процессы точного копирования молекул (репликация), так и процессы, которые влияют на саму структуру, несущую информацию (репарация и рекомбинация). Именно эти процессы мы и рассмотрим в данной главе.
Метаболизм ДНК должен происходить идеально точно. Химические реакции присоединения одного нуклеотида к другому при репликации ДНК элегантны и на удивление просты. Сложности, как мы увидим далее, в ферментативном аппарате, обеспечивающем точность передачи генетической информации. Ошибки, возникающие при синтезе ДНК, могут привести к серьезным последствиям, причем не только потому, что они изменяют или нарушают функцию определенного гена, но и потому, что такое изменение передается по наследству.
Ферменты, участвующие в синтезе ДНК. копируют молекулы ДНК, содержащие миллионы оснований. Они проделывают это с необыкновенной точностью и скоростью, несмотря на то что ДНК очень компактно упакована и связана с другими белками. Образование фосфодиэфирных связей между нуклеотидами в растущей цепи ДНК — только часть сложного процесса, в котором задействовано множество белков и ферментов.
Задача сохранения генетической информации лежит в основе процесса репарации ДНК. В гл. 8 (т. 1) подробно описана чувствительность ДНК ко многим разрушительным воздействиям. Такие воздействия происходят нечасто, но они играют очень важную роль, поскольку устойчивость организмов к изменениям последовательности ДНК очень низкая. ДНК — единственная макромолекула, для которой предусмотрена система репарации; количество, разнообразие и сложность механизмов репарации отражает многообразие пагубных для ДНК воздействий.
Клетки могут перестраивать хранящуюся в них генетическую информацию в процессах под общим названием «рекомбинация». Казалось бы, процесс рекомбинации подрывает принцип первостепенного значения стабильности и целостности генетической информации. Однако на самом деле большинство перестроек ДНК играет конструктивную роль в поддержании целостности генома, специфическим образом влияя на репликацию и репарацию ДНК и расхождение хромосом.
В этой главе особое внимание уделяется ферментам метаболизма ДНК. Они заслуживают внимательного изучения не только ввиду их большого биологического значения и чисто научного интереса, но и в связи с их возрастающей ролью в качестве лекарственных препаратов в медицине и в качестве инструментов в широком круге современных биохимических технологий. Многие ценные открытия в области метаболизма ДНК были сделаны на клетках Escherichia coli, поэтому для объяснения основ метаболизма обычно в качестве примера служат хорошо известные ферменты этой бактерии. Беглый взгляд на некоторые важные гены на генетической карте Е. coli (рис. 25-1) позволяет представить себе сложность ферментных систем, участвующих в метаболизме ДНК.
Рис. 25-1. Хромосомная карта Escherichia coli. Показано расположение генов, кодирующих многие важные для метаболизма ДНК белки. Количество известных генов, участвующих в метаболизме ДНК, свидетельствует о сложности этих процессов. Числа от 0 до 100 внутри кольцевой хромосомы соответствуют генетическим единицам измерения, называемым минутами. Каждая минута соответствует отрезку молекулы ДНК длиной примерно 40 000 п. н. Трехбуквеные названия генов обычно отражают некоторые аспекты их функции, к примеру, mut — мутагенез; dna — репликация ДНК; pol — ДНК-полимераза; rро — РНК-полимераза; uvr — устойчивость к УФ; rес — рекомбинация; dam — метилирование аденина; lig — ДНК-лигаза; Теr — терминация репликации; ori — точка начала репликации (у Е. coli это oriC, как на рисунке).
Прежде чем перейти к детальному изучению процесса репликации, сделаем короткое отступление и поговорим о принятых сокращениях названий бактериальных генов и белков, поскольку со многими из них нам предстоит встретиться в этой и последующих главах. Аналогичные договоренности используются и в обозначениях эукариотических генов, хотя конкретное сокращение может зависеть от вида организма, и единого правила для всех эукариотических систем не существует.
Ключевые договоренности.
Бактериальные гены обычно обозначают тремя строчными буквами курсивом, и, как правило, название отражает функцию этих генов. Например, dna, uvr и rес гены означают репликацию ДНК, устойчивость к поражающему действию УФ-облучения и рекомбинацию соответственно. Если несколько генов отвечают за одну и ту же функцию, они дополнительно обозначаются буквами А, В, С и т. д., например, dnaA, dnaB, dnaQ, что обычно указывает на порядок их открытия, а не порядок участия в цепи реакций. ■
Использование сокращений в названиях белков менее очевидно. В ходе генетических исследований обычно выделяют и характеризуют белковые продукты каждого гена. Многие бактериальные гены были идентифицированы и названы до того, как выяснилось значение их белковых продуктов. Иногда оказывается, что продукт гена — это уже известный белок, и его приходится переименовывать. Однако часто бывает, что продукт гена еще неизвестен и обладает активностью, которую нельзя описать обычным названием фермента.
Ключевые договоренности.
Бактериальные белки часто сохраняют названия соответствующих генов. Названия белков Е. coli пишутся прямым шрифтом с заглавной буквы, например, белковые продукты генов dnaA и rесА называются DnaA и RecA соответственно. ■
25.1. Репликация ДНК
Задолго до установления структуры ДНК ученых удивляла способность организмов воссоздавать самих себя и способность клеток образовывать много идентичных копий крупных и сложных макромолекул. Рассуждения на эту тему концентрировались вокруг концепции матрицы — структуры, которая позволяет молекулам соединяться в определенном порядке и образовывать макромолекулу с уникальной последовательностью и функцией. В 1940-е гг. уже сложилось представление о том, что носителем генетической информации является ДНК, но только когда Джеймс Уотсон и Фрэнсис Крик установили ее структуру, стало понятно, каким образом ДНК играет роль матрицы для репликации и передает генетическую информацию: дело в том, что одна ее цепь комплементарна другой. Основания образуют пары по определенным правилам, и каждая цепь является матрицей для новой цепи с предсказуемой комплементарной последовательностью (см. рис. 8-14, 8-15 в т. 1).
Было доказано, что основные свойства процесса репликации ДНК и каталитические механизмы этого процесса в значительной степени идентичны у всех видов организмов. Именно это единство механизмов мы постараемся подчеркнуть в нашем обсуждении, продвигаясь от общих основ процесса репликации к ферментам репликации Е. coli и, наконец, к репликации эукариот.
Основные принципы репликации ДНК
В ранних исследованиях репликации бактериальной ДНК и ее ферментов было установлено несколько базовых принципов, на которых основан синтез ДНК у всех живых существ.
Репликация ДНК полуконсервативна.
Каждая цепь ДНК служит матрицей для синтеза новой цепи, при этом образуются две новые двухцепочечные молекулы ДНК, каждая из которых состоит из одной новой и одной старой цепей. Поэтому процесс называется полуконсервативной репликацией.
Уотсон и Крик выдвинули гипотезу полуконсервативной репликации вскоре после публикации своей статьи о структуре ДНК с 1953 г.; в 1957 г. эта гипотеза была подтверждена Мэтью Мезельсоном и Франклином Сталем, которые искусно провели следующие эксперименты. Мезельсон и Сталь культивировали клетки Е. coli на протяжении многих генераций в среде, в которой единственный источник азота (NH4Cl) содержал тяжелый изотоп азота 15N вместо наиболее распространенного легкого изотопа 14N. Плотность выделенной из этих клеток ДНК примерно на 1% больше, чем у обычной [14N] ДНК (рис. 25-2, а). Несмотря на столь небольшое различие, смесь тяжелой [15N] ДНК и легкой [14N] ДНК можно разделить центрифугированием в градиенте плотности хлорида цезия.
Рис. 25-2, а — несколько поколений клеток выращивали в среде, содержащей только тяжелый азот 15N, поэтому в ДНК этих клеток содержался исключительно азот 15N, что показано в виде одной (синей) полосы, образующейся при центрифугировании в градиенте плотности CsCl. б — клетки переносили на среду, содержащую только легкий азот 14N, и после первого деления клеток выделяли ДНК, которая в градиенте хлорида цезия образовывала более высокую (фиолетовую) полосу, в — после второго цикла репликации ДНК разделялась на две полосы: гибридную (фиолетовую) и еще более легкую (красную), содержащую только ДНК с 14N, что подтверждало полу- консервативный характер репликации.
Клетки Е. coli, выращенные на среде с 15N, переносили на свежую питательную среду, содержащую только изотоп 14N, где клетки продолжали расти до тех пор, пока численность популяции не удваивалась. ДНК, выделенную из этой первой генерации клеток, центрифугировали в градиенте CsCl и наблюдали единственную полосу, положение которой свидетельствовало о том, что двухспиральная ДНК дочерних клеток представляет собой гибрид, содержащий одну новую цепь с 14N и одну родительскую цепь с 15N (рис. 25-2, б).
Представленные результаты опровергли альтернативную теорию консервативной репликации, согласно которой одна дочерняя молекула ДНК содержит две вновь синтезированные цепи, а другая дочерняя молекула — две родительские цепи; если бы эта теория была верна, в эксперименте Мезельсона -Сталя не было бы гибридной ДНК. Гипотеза полуконсервативной репликации дополнительно подтверждалась на следующем этапе эксперимента (рис. 25-2, в). После второго деления и удвоения клеток на среде с 14N из них была выделена ДНК, разделившаяся в градиенте хлорида цезия на две полосы: одна по плотности соответствовала легкой ДНК, а другая — гибридной ДНК. полученной после первого деления клеток.
Репликация начинается в точке начала репликации и обычно идет в двух направлениях.
После доказательства полуконсервативного механизма репликации возникло множество вопросов. Полностью ли раскручивается родительская ДНК до начала репликации каждой цепи? Начинается ли репликация в случайной или в какой-то особой точке? После инициации в какой-то точке репликация происходит в одном направлении или в двух?
В ранних исследованиях Джона Кэрнса, проведенным с помощью метода авторадиографии, удалось доказать, что репликация — высококоординированный процесс, в котором родительские цепи одновременно расплетаются и реплицируются. Кэрнс получил радиоактивную ДНК Е. coli, для чего культивировал эту бактерию на среде с меченным тритием (3Н) тимидином. ДНК была аккуратно выделена, покрыта слоем фотографической эмульсии и оставлена так на несколько недель; за это время радиоактивный тимидин оставил в фотоэмульсии «следы» — зерна серебра, создав фотоотпечаток молекулы ДНК. По этим отпечаткам видно, что интактная хромосома Е. coli представляет собой единую гигантскую кольцевую структуру длиной 1,7 мм. В радиоактивной ДНК, выделенной из клеток во время репликации, кроме того, была обнаружена дополнительная петля (рис. 25-3, а). Кэрнс сделал вывод, что, петля образуется в результате формирования двух радиоактивных дочерних цепей, каждая из которых комплементарна родительской пени. На одном или на обоих концах петли движется репликативная вилка, в которой родительская ДНК раскручивается, позволяя реплицироваться разделившимся цепям. Согласно полученным Кэрнсом данным, обе цепи ДНК реплицируются одновременно, а различные варианты его эксперимента (рис. 25-3, б) показали, что репликация бактериальных хромосом двунаправленная: на обоих концах петли расположены активные репликативные вилки.
Рис. 25-3. Выявление двунаправленного характера репликации ДНК. а — во время репликации кольцевой хромосомы образуется структура, напоминающая греческую букву тета (θ), так как обе нити реплицируются одновременно (новая нить показана красным цветом). б — репликация может происходить в одном или в обоих направлениях, что можно установить с помощью авторадиографии: при внесении 3Н на короткий период непосредственно перед прекращением реакции метка (отмечена красным) обнаруживается в одной или в двух репликативных вилках соответственно. Так была продемонстрирована двунаправленность репликации у Е. coli, Bacillus subtilis и других бактерий. На авторадиограмме показан репликационный «глазок» в ДНК В. subtilis. Самая большая плотность зерен серебра (стрелки) наблюдается именно в тех двух местах, где происходит репликация. Не подвергшаяся репликации часть хромосомы, находящаяся вне глазка, не содержит метки и поэтому невидима.
Чтобы выяснить, образуются ли репликативные вилки в каком-то определенном участке ДНК, необходимо было установить по всей длине молекулы некие реперные точки. Это было сделано с помощью метода денатурирующего картирования, разработанного Россом Инманом с коллегами. С помощью хромосомы бактериофага λ длиной 48 502 п. и. было показано, что можно вызвать избирательную денатурирацию ДНК в последовательностях с необычно высоким содержанием пар А = Т, получая воспроизводимую картину одноцепочечных глазков (см. рис. 8-28 в т. 1). Таким же образом можно частично денатурировать выделенную ДНК, содержащую репликативные петли. Это позволяет измерить и картировать положение и движение репликативных вилок, используя денатурированные области в качестве реперных точек. С помощью данного метода было показано, что в этой системе репликативные петли всегда возникают в особых точках, которые были названы точками начала репликации (ориджинами). Получил подтверждение и выявленный ранее двунаправленный характер репликации. У кольцевых молекул ДНК две репликативные вилки встречаются в точке, расположенной напротив точки начала репликации. Специфические точки начала репликации с тех пор были идентифицированы и охарактеризованы у бактерий и низших эукариот.
Синтез ДНК наполовину прерывистый и проходит в направлении 5’—> 3′.
Новая цепь ДНК всегда синтезируется в направлении 5′ —> 3′, т. е. приращение ДНК происходит со стороны свободной ОН-группы на 3′-конце (строение 5′- и 3′-концов цепи ДНК см. на рис. 8-7 в т. 1). Поскольку две цепи ДНК антипараллельны, цепь, служащая в качестве матрицы, считывается с 3’-конца в направлении к 5′-концу.
Но если синтез ДНК всегда происходит в направлении 5′ —> 3′, как могут обе цепи синтезироваться одновременно? Если обе цепи синтезируются непрерывно, пока движется репликативная вилка, одна цепь должна синтезироваться в направлении 3′ —> 5′. Эту проблему в 1960-х гг. разрешил Рейджи Оказаки с коллегами. Они обнаружили, что одна из новых цепей ДНК синтезируется в виде коротких отрезков, которые теперь называются фрагментами Оказаки. В результате их исследований стало ясно, что одна цепь синтезируется непрерывно, а вторая — прерывисто (рис. 25-4). Направление синтеза непрерывной, или лидирующей, цепи совпадает с направлением движения репликативной вилки. Направление синтеза прерывистой, или отстающей, цепи противоположно направлению движения вилки. Фрагменты Оказаки различаются по длине от нескольких сотен до нескольких тысяч нуклеотидов в зависимости от типа клеток. Ниже мы увидим, что синтез лидирующей и отстающей цепей четко координируется.
Рис. 25-4. Различия цепей ДНК на репликативной вилке. Новая цепь ДНК (красная) всегда синтезируется в направлении 5′ —> 3′. Матрица считывается в противоположном направлении. 3′ —> 5′. Лидирующая цепь непрерывно синтезируется по направлению движения репликативной вилки. Другая цепь, отстающая, синтезируется прерывисто в виде коротких отрезков (фрагментов Оказаки) в направлении, противоположном тому, в котором движется репликативная вилка. Фрагменты Оказаки сшиваются ДНК-лигазой. У бактерий длина фрагментов Оказаки составляет примерно от 1000 до 2000 нуклеотидов. В клетках эукариот они короче: от 150 до 200 нуклеотидов.
ДНК разрушается нуклеазами
Чтобы объяснить энзимологию процесса репликации ДНК, сначала рассмотрим ферменты, которые разрушают, а не синтезируют ДНК. Эти ферменты называют нуклеазами или ДНКазами, если они обладают большей специфичностью к ДНК, чем к РНК. Каждая клетка содержит несколько разных нуклеаз, принадлежащих к двум большим классам: экзонуклеазы и эндонуклеазы. Экзонуклеазы расщепляют нуклеиновые кислоты с одного конца молекулы. Многие из них работают только в направлении 5′ —> 3′ или только в направлении 3′ —> 5′, удаляя нуклеотиды либо с 5′-конца, либо с 3′-конца одной цепи двухцепочечной молекулы нуклеиновой кислоты или одноцепочечной ДНК. Эндонуклеазы начинают расщепление нуклеиновой кислоты со специфических внутренних сайтов, расщепляя цепь на более мелкие фрагменты. Некоторые экзонуклеазы и эндонуклеазы расщепляют только одноцепочечные ДНК. Существует несколько важных классов эндонуклеаз, которые разрушают только определенные нуклеотидные последовательности (например, эндонуклеазы рестрикции, которые имеют очень большое значение в биотехнологии; см. гл. 9, рис. 9-2 вт. 1). В этой и в последующих главах мы встретим много типов нуклеаз.
ДНК синтезируется ДНК-полимеразами
Поиск фермента, который может синтезировать ДНК, начался в 1955 г. Артуру Корнбергу с коллегами удалось очистить и охарактеризовать ДНК-полимеразу из клеток Е. coli. Этот фермент, состоящий из одного полипептида, теперь называют ДНК-полимеразой I (Мr = 103 000; кодируется геном роlА). Гораздо позже исследователи обнаружили, что Е. coli содержит еще как минимум четыре другие ДНК-полимеразы, описанные ниже.
При детальном изучении ДНК-полимеразы 1 были установлены общие для всех ДНК-полимераз характеристики процесса синтеза ДНК. В основной реакции происходит перенос фосфорильных групп. При этом 3′-гидроксильная группа нуклеотида на 3′-конце растущей цепи выступает в роли нуклеофила, атакующего α-фосфор присоединяющегося дезоксинуклеозид-5′-трифосфата (рис. 25-5). В результате высвобождается неорганический пирофосфат. Основная реакция выглядит следующим образом:
где dNMP и dNTP — дезоксинуклеозид-5v-монофосфат и дезоксинуклеозид-5′-трифосфат соответственно. Реакция происходит с минимальным изменением свободной энергии, поскольку одна фосфодиэфирная связь образуется из менее стабильного фосфоангидрида. Нековалентные стэкинговые взаимодействия оснований и их спаривание обеспечивают дополнительную стабилизацию удлиняющейся ДНК по сравнению со свободными нуклеотидами. Кроме того, синтез ДНК приводит к высвобождению в клетке 19 кДж/моль энергии в результате последующего гидролиза пирофосфата под действием фермента пирофосфатазы (с. 38 в т. 2).
Рис. 25-5. МЕХАНИЗМ РЕАКЦИИ. Элонгация цепи ДНК. а — ДНК-полимераза I нуждается в одной неспаренной цепи в качестве матрицы и в праймере, несущем на 3′-конце свободную гидроксильную группу, к которой присоединяется новое нуклеотидное звено. Каждый присоединяющийся нуклеотид выбирается по принципу комплементарности по отношению к соответствующему нуклеотиду в матричной цепи. Продукт реакции вновь содержит свободную 3′-гидроксильную группу, позволяющую присоединять следующий нуклеотид. б — для катализа, вероятно, требуется два иона Мg2+, координированных с двумя фосфатными группами присоединяющегося нуклеозидтрифосфата и с тремя остатками Asp, два из которых консервативны у всех ДНК-полимераз. Ион Мg2+, изображенный справа, облегчает атаку 3′-гидроксильной группы праймера на α-фосфат нуклеозидтрифосфата другой ион Мg2+ способствует удалению пирофосфата. Оба иона стабилизируют структуру пятикоординационного переходного состояния. По схожему механизму действует и РНК-полимераза (см. рис. 26-1, б).
Еще в ранних исследованиях ДНК-полимеразы I удалось определить два основных условия, необходимых для полимеризации ДНК. Во-первых, все ДНК-полимеразы нуждаются в матрице. Реакция полимеризации происходит на матрице ДНК согласно правилам комплементарности оснований, установленным Уотсоном и Криком: если в матрице присутствует гуанин, к новой цепи присоединяется дезоксинуклеотид цитозина и т. д. Это открытие имеет очень важное значение не только потому, что оно объясняет химическую основу точной полуконсервативной репликации ДНК, но и потому, что это был первый пример использования матрицы для проведения реакции биосинтеза.
Во-вторых, полимеразы нуждаются в праймере. Праймер — это участок цепи (комплементарный матрице) со свободной З’-гидроксильной группой, к которой может присоединиться нуклеотид. Иначе говоря, перед началом синтеза часть новой цепи уже должна существовать: все ДНК-полимеразы могут присоединять нуклеотиды только к уже существующей цепи. Многие праймеры представляют собой олигонуклеотиды РНК, а не ДНК; специальные ферменты синтезируют праймеры там и тогда, где и когда они требуются.
После присоединения нуклеотида к растущей цепи ДНК ДНК-полимераза либо диссоциирует, либо продвигается вдоль матрицы и присоединяет следующий нуклеотид. Диссоциация и обратное присоединение полимеразы может ограничивать среднюю скорость полимеризации — процесс обычно протекает быстрее, если полимераза присоединяет нуклеотиды, не покидая матрицы. Среднее число нуклеотидов, присоединенных до диссоциации полимеразы, определяет ее процессивность. ДНК-полимеразы существенно различаются по процессивности; некоторые присоединяют всего несколько нуклеотидов до диссоциации, другие присоединяют многие тысячи.
Репликация — очень точный процесс
Репликация осуществляется с исключительной точностью. У Е. coli происходит всего одна ошибка на каждые 109-1010 присоединенных нуклеотидов. Для хромосомы Е. coli, размер которой составляет примерно 4,6 • 106 п. н., это означает, что за 1000-10 000 циклов репликации случается лишь одна ошибка. Во время полимеризации выбор между правильным и неправильным нуклеотидом осуществляется не только по специфичности водородных связей между комплементарными основаниями в парах, но также по геометрии стандартных А = Т и G = C пар оснований (рис. 25-6). В активном центре ДНК-полимеразы I могут разместиться только пары оснований с правильной геометрией. Неподходящий нуклеотид может образовать водородные связи с нуклеотидом в матрице, но такая пара обычно не попадает в активный центр фермента. Это позволяет отбраковать неправильное основание до того, как сформируется фосфодиэфирная связь.
Рис. 25-6. Значение геометрии пар оснований для точности репликации ДНК. а — стандартные пары оснований А = Т и G = C очень схожи по геометрии, и, если активный центр фермента может вместить одну пару (синяя рамка), то обычно он вмещает и другую, б — геометрия аномальных пар отличается, и они не помещаются в активном центре ДНК-полимеразы I.
Однако, чтобы обеспечить высокую степень точности репликации, одной только точности реакции полимеризации недостаточно. Измерения in vitro показали, что ДНК-полимераза вставляет один неправильный нуклеотид на 104 — 105 правильных нуклеотидов. Иногда эти ошибки возникают из-за того, что основание кратковременно находится в нехарактерной таутомерной форме (см. рис. 8-9 в т. 1), что позволяет ему образовать водородные связи не с тем партнером. Число ошибок in vivo уменьшается благодаря дополнительным ферментативным механизмам.
Один механизм сокращения числа ошибок свойствен практически всем ДНК- полимеразами и заключается в независимой 3′ —> 5′-экзонуклеазной активности, благодаря которой каждый нуклеотид после присоединения проверяется дважды. Эта нуклеазная активность чрезвычайно чувствительна к аномальному спариванию оснований, что позволяет ферменту удалять ошибочно присоединенный нуклеотид (рис. 25-7). Если полимераза присоединила неправильный нуклеотид, происходит ингибирование ее перемещения к позиции, в которой должен присоединяться следующий нуклеотид. Эта пауза создает возможность для исправления ошибки. С помощью 3′ —> 5′-экзонуклеазной активности полимераза удаляет неправильный нуклеотид и продолжает удлинение цепи. Данная активность, называемая корректирующей, не просто обращение полимеразной реакции (уравнение 25-1), ведь в ней не участвует пирофосфат. Полимеразную и корректирующую активности ДНК-полимеразы можно измерить независимо. Корректирующая активность дополнительно увеличивает точность реакции полимеризации в 102-103раз. У мономерной ДНК-полимеразы I полимеразная и корректирующая активности локализованы в разных активных центрах в одном и том же полипептиде.
Рис. 25-7. Пример исправления ошибки благодаря 3′ —> 5′-экзонуклеазной активности ДНК-полимеразы I. При изучении структуры фермента было установлено, что при ориентации фермента по ходу движения ДНК- центр его экзонуклеазной активности локализован впереди центра полимеразной активности. Ошибочное спаривание (здесь ошибка С-А) препятствует транслокации ДНК-полимеразы I в следующее положение. Фермент скользит назад, исправляет ошибку благодаря своей 3′ —> 5′-экзонуклеазной активности, а затем возобновляет полимеразную активность в направлении 5′ —> 3′.
При тщательном подборе оснований в сочетании с исправлением ошибок ДНК-полимераза совершает примерно одну ошибку на 106-108 присоединенных оснований. В реальности измеряемая точность репликации Е. coli еще выше. Дополнительная точность обеспечивается независимой ферментативной системой, которая заменяет аномальные пары оснований, оставшиеся после репликации. Данный процесс исправления ошибок, наряду с другими процессами репарации ДНК, описан в разд. 25.2.
У Е. coli не менее пяти ДНК-полимераз
Более 90% ДНК-полимеразной активности в экстрактах Е. coli связано с функцией ДНК-полимеразы I. Однако вскоре после выделения этого фермента в 1955 г. стали накапливаться сведения о том, что он не может осуществлять репликацию крупной хромосомы Е. coli. Во-первых, скорость, с которой фермент присоединяет нуклеотиды (600 нуклеотидов/мин), не соответствует скорости движения репликативной вилки в бактериальной клетке (скорость движения репликативной вилки, как минимум, в 100 раз выше). Во- вторых, ДНК-полимераза I обладает относительно низкой процессивностью. В-третьих, генетические исследования показали, что в репликации участвуют многие гены и, следовательно, многие белки: очевидно, что ДНК-полимераза I действует не в одиночку. В-четвертых, и это особенно важно, в 1969 г. Джон Кэрнс выделил бактериальный штамм с поврежденным геном ДНК-полимеразы I, с которого синтезировался неактивный фермент. Хотя этот штамм был чрезвычайно чувствителен к повреждениям ДНК, тем не менее он был жизнеспособен!
Поиск других ДНК-полимераз привел к открытию в начале 1970-х гг. ДНК-полимеразы II и ДНК-полимеразы III у Е. coli. ДНК-полимераза II участвует в одном из механизмов репарации ДНК (разд. 25.3). ДНК-полимераза III — главный фермент репликации у Е. coli. Свойства трех ДНК-полимераз Е. coli сравниваются в табл. 25-1. ДНК-полимеразы IV и V, идентифицированные в 1999 г., принимают участие в необычном варианте репарации ДНК (разд. 25.2).
Таблица 25-1. Сопоставление ДНК-полимераз Е. coli
|
ДНК-полимер |
аза |
||
|
I |
II |
III |
|
|
Структурный гена |
polA |
роlB |
polC (dnaE) |
|
Субъединицы (количество разных типов) |
1 |
7 |
≥10 |
|
Относительная молекулярная масса (Mr) |
103 000 |
88 000б |
791 500 |
|
3′ —> 5′-экзонуклеаза (коррекция ошибок) |
Да |
Да |
Да |
|
5′ —> 3′-экзонуклеаза |
Да |
Нет |
Нет |
|
Скорость полимеризации (нуклеотиды/с) |
16-20 |
10 |
250- 1000 |
|
Процессивность (число нуклеотидов, присоединенных до диссоциации полимеразы) |
3-200 |
1500 |
≥500 000 |
а Для субъединичных ферментов назван ген, кодирующий субъединицу с полимеразной активностью. Ген dnаE в настоящее время называется polC.
б Только субъединица, отвечающая за полимеризацию. В составе ДНК-полимеразы II есть те же субъединицы, что и у ДНК-полимеразы III: (β-, y-, δ-, δ’-, χ- и Ψ-субъединицы (см. табл. 25-2).
ДНК-полимераза I, таким образом, не главный фермент в репликации; она выполняет функцию проверки и поддержания порядка во время репликации, рекомбинации и репарации. Особые функции полимеразы дополняются ее 5′ —> 3′-экзо- нуклеазной активностью. Эта активность, отличная от корректирующей 3′ —> 5′-экзонуклеазной активности (рис. 25-7), локализована в структурном домене, который может быть отщеплен от фермента в условиях мягкой обработкой протеазами. При удалении 5′ —> 3′-экзонуклеазного домена остающийся фрагмент (Мr = 68 000), называемый большим фрагментом, или фрагментом Клёнова (рис. 25-8), сохраняет полимеразную и корректирующую активность. Интактная ДНК-полимераза I благодаря своей 5′ —> 3′-экзонуклеазной активности в ходе так называемой ник-трансляции может заменить участок ДНК (или РНК), связанный с матричной цепью (рис. 25-9). Большинство других ДНК-полимераз не имеют 5′ —> 3′-экзонуклеазной активности.
Рис. 25-8. Большой фрагмент ДНК-полимеразы I (фрагмент Клёнова). Эта полимераза широко распространена у бактерий. Фрагмент Клёнова, образующийся в результате протеолитической обработки полимеразы, сохраняет полимеразную и корректирующую активности. Представленный на рисунке фрагмент Клёнова получен из термофильной бактерии Bacillus stearothermophilus (PDB ID 3BDP). Активный центр полимеразы находится глубоко в щели около дальнего конца связанной ДНК (синяя; темно-синим цветом изображена матричная цепь).
Рис. 25-9. Ник-трансляция. В ходе этого процесса участок РНК или ДНК, спаренный с матричной ДНК, одновременно разрушается под действием 5′ —> 3′-экзонуклеазной активности ДНК-полимеразы I и заменяется на новый под действием полимеразной активности того же фермента. Эта функция реализуется в процессе репарации ДНК и при удалении РНК-праймеров в ходе репликации (оба процесса описаны ниже). Цепь нуклеиновой кислоты (ДНК или РНК), подлежащая удалению, показана зеленым, новая цепь — красным. Синтез ДНК начинается с ника (разрыва фосфодиэфирной связи, освобождающего 3′-гидроксильную и 5′-фосфатную группы). Полимераза I удлиняет нематричную цепь ДНК и перемещает ник вдоль ДНК. Этот процесс называется ник-трансляцией. В месте диссоциации ДНК-полимеразы ник сохраняется и позднее зашивается другим ферментом.
ДНК-полимераза III устроена намного сложнее, чем ДНК-полимераза I; она состоит из субъединиц десяти разных типов (табл. 25-2). Ее полимеразная и корректирующая активности связаны с α- и ε-субъединицами соответственно. При объединении θ-субъединицы с α- и ε-субъединицами образуется корфермент, который уже может осуществлять синтез ДНК, но с ограниченной процессивностью. Два кор-фермента полимеразы могут связываться друг с другом посредством еще одного субъединичного комплекса, называемого погрузчиком зажима, или y-комплексом, состоящим из пяти субъединиц четырех разных типов Ƭ2yδδ’. Кор-ферменты связаны через Ƭ-субъединицы. Две дополнительные единицы χ и Ψ, присоединены к погрузчику зажима. Полный набор из 13 белковых субъединиц (девяти разных типов) называется ДНК-полимеразой III* (рис. 25-10, а).
Таблица 25-2. Субъединицы ДНК-полимеразы III E. coli
а y-Субъединицу кодирует часть гена Ƭ-субъединицы, так что 66% Ƭ-субъединицы с N-конца совпадают по аминокислотной последовательности с y-субъединицей. y-Субъединица образуется в результате смещения рамки считывания при трансляции (см. рис. 27-9), что приводит к преждевременной остановке трансляции.
ДНК-полимераза III* способна полимеризовать ДНК, но с намного меньшей процессивностью, чем нужно для организованной репликации целой хромосомы. Необходимое увеличение процессивности обеспечивается присоединением четырех β-субъединиц с образованием полного фермента ДНК-полимеразы III. β-Субъединицы объединяются в пары и формируют структуры в форме бубликов, которые охватывают молекулы ДНК, как зажимы (рис. 25-10, б). Каждый димер связывается с кор-ферментом полимеразы III* (один димерный зажим на кор-фермент) и во время репликации скользит вдоль ДНК. Скользящий β-зажим предупреждает диссоциацию ДНК-полимеразы III из комплекса с ДНК, резко увеличивая процессивность до 500 000 нуклеотидов и более (табл. 25-1).
Рис. 25-10. ДНК-полимераза III. а — строение бактериальной ДНК-полимеразы III. Два центральных домена (кор-ферменты), состоящие из субъединиц α, ε и θ, связаны с y-комплексом (погрузчиком зажима), состоящим из пяти субъединиц Ƭ2yδδ’. Субъединицы y и Ƭ кодируются одним и тем же геном. Субъединица у представляет собой укороченную субъединицу Ƭ: субъединица Ƭ содержит домен, идентичный у, и дополнительный сегмент, взаимодействующий с кор-ферментом. Две другие субъединицы χ и Ψ (не показаны) ДНК-полимеразы III* также связаны с y-комплексом. Два β-зажима присоединяются к двум кор-ферментам; каждый зажим — это димер β-субъединицы. Комплекс взаимодействует с DnaB- хеликазой через Ƭ-субъединицу. б — две β-субъединицы полимеразы III Е. coli образуют зажим в виде кольца, которое окружает ДНК. Зажим скользит по молекуле ДНК, увеличивая процессивность холофермента ДНК-полимеразы III до 500 000 нуклеотидов и более, не позволяя ДНК диссоциировать. Вид снизу демонстрирует две β-субъединицы, изображенные в виде лент голубого и серого цвета, окружающих СРК-модель молекулы ДНК. Вид сбоку демонстрирует контур поверхности β-субъединиц (серого цвета), которые окружают двойную спираль ДНК (голубой и синий цвет), изображенную в виде стрежневой модели (по PDB ID 2PОL).
В репликации ДНК участвует множество ферментов и белковых факторов
Для репликации Е. coli нужна не только ДНК-полимераза, но также не менее 20 различных ферментов и белков, каждый из которых выполняет определенную задачу. Весь этот комплекс был назван ДНК-репликативной системой, или реплисомой. Сложность ферментативного аппарата репликации связана со структурой ДНК и с требованиями высокой точности синтеза. Основные классы ферментов репликации перечислены ниже в соответствии с теми функциями, которые они выполняют.
Чтобы обеспечить доступ к цепям ДНК, которые служат в качестве матрицы, две родительские цепи нужно разделить. Обычно этот процесс осуществляют хеликазы — ферменты, которые движутся вдоль ДНК и разделяют ее цепи, затрачивая при этом энергию АТР. Разделение цепей создает топологическое напряжение в спиральной структуре ДНК (см. рис. 24-12), которое снимается топоизомеразами. Разделенные цепи стабилизируются ДНК-связывающими белками. Как уже было сказано, прежде чем ДНК-полимеразы смогут начать синтез ДНК, на матрице должны находиться праймеры — обычно это короткие сегменты РНК, синтезированные ферментами праймазами. Позднее РНК-праймеры удаляются и заменяются на ДНК; у Е. coli это действие — одна из основных функций ДНК-полимеразы I. После вырезания РНК-праймера и встраивания ДНК в последовательности ДНК сохраняется ник в форме разорванной фосфодиэфирной связи. Эти ники зашивают ДНК- лигазы. Все перечисленные процессы нуждаются в координации и регуляции, которые хорошо исследованы в системе Е. coli.
Репликация хромосомы E. coli происходит постадийно
В синтезе молекулы ДНК можно выделить три стадии: инициация, элонгация и терминация, которые различаются как по химической сути, так и по составу ферментов. В этой и двух последующих главах рассматриваются именно эти три общие стадии синтеза крупных информационных полимеров ДНК, РНК и белков, а также специфические особенности каждого биосинтетического пути. Описываемые далее события базируются на информации, первоначально полученной в экспериментах in vitro на очищенных белках Е. coli, хотя принципы репликации весьма консервативны во всех системах.
Инициация.
Точка начала репликации Е. coli (oriC) состоит из 245 п. н.; она содержит элементы последовательности, консервативные во многих бактериальных точках начала репликации. Стандартный набор консервативных последовательностей показан на рис. 25-11. Наибольший интерес представляют два типа последовательностей: пять повторов размером по 9 п. н. (R-сайты) — участки связывания ключевого инициаторного белка DnaA, а также А = Т-богатый участок, называемый ДНК-расплетающим элементом DUE (от англ. DNA unwinding element). Кроме того, существуют три дополнительных участка связывания DnaA (I -сайты) и участки связывания белков IHF (от англ. integration hose factor) и FIS (от англ. factor for inversion stimulation). Эти два белка необходимы для некоторых реакций рекомбинации, описанных ниже в данной главе, а их названия отражают их функции. Еще один ДНК-связывающий белок HU (гистоноподобный бактериальный белок, ранее называвшийся фактором U) также участвует в процессе, но не имеет специфического участка связывания.
Рис. 25-11. Расположение последовательностей в точке начала репликации Е. coli (oriC). Показаны консенсусные последовательности (см. с. 156 в т. 1) ключевых повторов. N обозначает любой из четырех нуклеотидов. Горизонтальные стрелки показывают ориентацию нуклеотидных последовательностей (стрелки слева направо указывают на последовательность в верхней цепи, а справа налево — в нижней). Участки FIS и IHF — сайты связывания белков, описанных в тексте. С R-сайтами связан белок DnaA. Сайты I — дополнительные участки связывания DnaA (у них другие последовательности). DnaA взаимодействует с ними только в комплексе с АТР.
В инициации репликации участвуют по меньшей мере 10 разных ферментов и белков (см. табл. 25-3). Они расплетают спираль ДHК в точке начала репликации и собирают препраймирующий комплекс для последующих реакций. Ключевым элементом в процессе инициации выступает белок DnaA — входит в белковое семейство ААА+ АТРаз (от англ. ATPases associated with diverse cellular activities). Многие AAA+ АТРазы, включая DnaA, образуют олигомерные структуры и сравнительно медленно гидролизуют АТР. Гидролиз АТР служит переключателем, опосредующим переходы одной формы белка в другую. В случае DnaA ATP-связанная форма активна, а ADP-связанная форма неактивна.
Восемь молекул белка DnaA, каждая из которых связана с АТР, образуют спиральный комплекс, охватывающий R- и I-сайты в oriC (рис. 25-12). DnaA имеет более высокое сродство к R-сайтам, чем к 1-сайтам, и этот белок связывается с R-сайтами одинаково хорошо как в АТР-, так и в ADP-форме. Напротив, 1-сайты, в которых связывается только ATP-связанный DnaA, позволяют различать активную и неактивную форму DnaA. Молекула ДНК плотно закручивается в правую сторону вокруг этого комплекса, образуя положительную суперспираль (см. гл. 24). Возникшее в соседних участках ДНК напряжение вызывает денатурацию А = Т-богатого участка DUE. Кроме того, образующийся в точке начала репликации комплекс также содержит несколько ДНК-связываюших белков — HU, IHF и FIS, которые облегчают изгиб ДНК.
Рис. 25-12. Модель инициации репликации в точке начала репликации Е. coli (оri’С). Восемь связанных с АТР молекул белка DnaA присоединяются к точке начала репликации в R- и I-сайтах (см. рис. 25-11). ДНК закручивается вокруг этого комплекса с образованием правозакрученной спирали. Богатый остатками А = Т участок DUE денатурирует из-за напряжения, возникающего в цепи в результате связывания DnaA. Образованию спирального комплекса с DnaA способствуют белки HU, IHF и FIS (здесь не показаны, так как подробно их структура и роль еще не определены). Гексамеры белка DnaB связываются с каждой цепью ДНК с помощью белка DnaC. Благодаря своей хеликазной активности белок DnaB раскручивает ДНК, подготавливая ее для посадки праймера и синтеза ДНК.
Затем другая ААА+ АТРаза, DnaC, загружает белок DnaB на разделенные цепи ДНК в денатурированном участке. Гексамер, состоящий из молекул DnaC, каждая из которых связана с АТР, образует прочный комплекс с гексамерной кольцевой DnaB-хеликазой. При взаимодействии DnaC с DnaB кольцо DnaB раскрывается, чему также способствует дальнейшее взаимодействие между DnaB и DnaA. Два кольцевых гексамера DnaB связываются с DUE — по одному на каждую нить ДНК. Связанная с DnaC молекула АТР гидролизуется, что приводит к высвобождению DnaC, a DnaB остается связанным с ДНК.
Присоединение хеликазы DnaB — ключевой момент в инициации репликации. Далее репликативная хеликаза DnaB перемещается вдоль одноцепочечной ДНК в направлении 5′ —> 3′, раскручивая ДНК по мере продвижения. Таким образом. связанные с двумя пенями ДНК молекулы хеликазы DnaB перемещаются в противоположных направлениях, создавая две репликативные вилки. Все другие белки репликативной вилки также напрямую или опосредованно связаны с DnaB. Холофермент ДНК-полимеразы III связан через Ʈ-субъединицы. Другие взаимодействия DnaB описаны ниже. В начале репликации, когда нити ДНК разделяются в области репликативной вилки, с ними связываются, стабилизируя их, многочисленные молекулы белка, связывающегося с одноцепочечной ДНК (single-stranded DNA-binding protein, SSB), а ДНК-гираза (ДНК- топоизомераза II) снимает топологическое напряжение, возникающее впереди вилки из-за раскручивания ДНК.
Инициация — единственная фаза репликации ДНК, про которую известно, что она регулируется, притом таким образом, что репликация происходит только один раз за клеточный цикл. Механизм регуляции пока еще точно не установлен. но в ходе генетических и биохимических исследований было выявлено несколько независимых элементов регуляции.
Как только с ДНК связывается ДНК-полимераза III, а также β-субъединицы (что свидетельствует о завершении фазы инициации), с β-субъединицами связывается белок Hda, который взаимодействует с DnaA и стимулирует гидролиз связанного с ним АТР. Белок Hda (название этого фермента означает «гомологичный DnaA») — еще одна ААА+ АТРаза, родственная DnaA. Гидролиз АТР приводит к разборке комплекса DnaA в точке начала репликации. Медленное высвобождение ADP и связывание новых молекул АТР замыкает цикл превращений между неактивной (со связанным ADP) и активной (со связанным АТР) формами белка, который длится от 20 до 40 мин.
На ход инициации репликации влияет метилирование ДНК и взаимодействия с бактериальной плазматической мембраной. ДНК в области оriС метилируется Dam-метилазой (от англ. DNA adenine methylation.; табл. 25-3), которая вводит метальную группу по положению N6 в остатке аденина в палиндромной последовательности (5’) GATC. Точка начала репликации оriС у Е. coli содержит особенно много повторов GATC — их 11 в последовательности из 245 п. н. тогда как в среднем во всей хромосоме Е. coli этот повтор встречается одни раз на 256 п. н.
Таблица 25-3. Белки, необходимые для инициации репликации в точке начала репликации E. coli
|
Белок |
Мr |
Число субъединиц |
Функция |
|
Белок DnaA |
52 000 |
1 |
Распознает последовательности в точке начала репликации; раскрывает цепи ДНК в специальных сайтах точки начала репликации |
|
Белок DnaB (хеликаза) |
300 000 |
6* |
Раскручивает ДНК |
|
Белок DnaC |
29 000 |
6* |
Связывает DnaB в точке начала репликации |
|
HU |
19 000 |
2 |
Гистоноподобный ДНК-связывающий белок; стимулирует инициацию |
|
FIS |
22 500 |
2* |
ДНК-связывающий белок; стимулирует инициацию |
|
IHF |
22 000 |
2 |
ДНК-связывающий белок; стимулирует инициацию |
|
Праймаза (белок DnaG) |
60 000 |
1 |
Синтезирует РНК-праймеры |
|
Белок, связывающий одноцепочечную ДНК (SSB) |
75 600 |
4* |
Связывает одноцепочечную ДНК |
|
ДНК-гираза (ДНК-топоизомераза II) |
400 000 |
4 |
Ослабляет торсионное напряжение, вызываемое раскручиванием ДНК |
|
Dam-метилаза |
32 000 |
1 |
Метилирует последовательность (5′)GATC в oriС |
* В данном случае субъединицы одинаковые.
Непосредственно после репликации ДНК оказывается наполовину метилированной; в родительских цепях последовательности оriС метилированы, а во вновь синтезированных — нет. Эти наполовину метилированные последовательности оriС изолированы за счет взаимодействия с плазматической мембраной (механизм неизвестен) и связывания с белком SeqA. Через некоторое время последовательность оriС отделяется от плазматической мембраны, SeqA диссоциирует, и Dam-метилаза полностью метилирует ДНК до того, как она вновь связывает DnaA в новом раунде репликации.
Элонгация.
Фаза элонгации репликации включает две связанные между собой операции: синтез лидирующей цепи и синтез отстающей цепи. Для синтеза обеих цепей важно присутствие в репликатвной вилке нескольких ферментов. Сначала исходная ДНК раскручивается ДНК-хеликазой, а возникающее топологическое напряжение снимается топоизомеразами. Затем белок SSB стабилизирует каждую отдельную цепь. С этого момента синтез лидирующей цени и синтез отстающей цепи кардинально различаются.
Синтез лидирующей цепи происходит проще: он начинается с синтеза праймазой (белок DnaG) короткого (от 10 до 60 нуклеотидов) РНК- праймера в точке начала репликации. Для осуществления данной реакции DnaG взаимодействует с хеликазой DnaB. причем праймер синтезируется в направлении, которое противоположно направлению движения хеликазы DnaB. На самом деле хеликаза DnaB перемещается вдоль цепи, которая становится отстающей при синтезе ДНК. Однако первый праймер, синтезированный при первом взаимодействии DnaG-DnaB, служит для запуска синтеза лидирующей цепи ДНК в противоположном направлении. Дезоксирибонуклеотиды добавляются к этому праймеру ДНК-полимеразой III, связанной с DnaB-хеликазой, расположенной на противоположной цепи ДНК. Синтез лидирующей цепи происходит непрерывно со скоростью, соответствующей скорости раскручивания ДНК в репликативной вилке.
Как уже было отмечено, отстающая цепь синтезируется в виде коротких фрагментов Оказаки (рис. 25-13, а). Сначала праймаза синтезирует РНК-праймер. Затем, как и в процессе синтеза лидирующей цепи, ДНК-полимераза III связывается с праймером и присоединяет дезоксирибонуклеотиды (рис. 25-13, б). На этом этапе синтез каждого фрагмента Оказаки кажется простым, но на самом деле это весьма сложный процесс. Сложность заключается в координации синтезов лидирующей и отстающей цепей: обе цепи синтезируются одним асимметричным димером ДНК-полимеразы III, что достигается при образовании петли ДНК из отстающей цепи (рис. 25-14), сближающей две точки полимеризации.
Рис. 25-13. Синтез фрагментов Оказаки, а — на определенном расстоянии праймаза синтезирует РНК-праймер для нового фрагмента Оказаки. Если рассматривать две параллельные матричные цепи, синтез отстающей цепи формально происходит в направлении, противоположном направлению движения вилки, б — каждый праймер удлиняется ДНК-полимеразой III. в — синтез ДНК продолжается до тех пор, пока полимераза не достигнет праймера предыдущего фрагмента Оказаки. Для возобновления процесса вблизи репликативной вилки синтезируется новый праймер.
Синтез фрагментов Оказаки на отстающей цепи происходит с участием удивительного ферментативного аппарата. DnaB-хеликаза и DnaG- праймаза образуют отдельный функциональный блок, праймосому, в составе репликативного комплекса. ДНК-полимераза III использует один набор кор-ферментов (коровая полимераза) для непрерывного синтеза лидирующей цепи, в то время как другой набор кор-ферментов осуществляет циклы синтеза фрагментов Оказаки на петле отстающей цепи. DnaB-хеликаза, находящаяся перед ДНК-полимеразой III, раскручивает ДНК в репликативной вилке (рис. 25-14, а), продвигаясь вдоль матрицы отстающей цепи в направлении 5′ —> 3′. ДНК-праймаза время от времени связывается с DnaB-хеликазой и синтезирует короткий РНК-праймер (рис. 25-14, б). Затем погрузчик зажима ДНК-полимеразы III закрепляет на праймере новый скользящий β-зажим (рис. 25-14, в). Когда синтез фрагмента Оказаки заканчивается, репликация приостанавливается, кор-фермент ДНК-полимеразы III отсоединяется от одного зажима (а также от законченного фрагмента Оказаки) и связывается с новым зажимом (рис. 25-14, г, б). Это инициирует синтез нового фрагмента Оказаки. Как отмечалось ранее, весь комплекс, ответственный за координированный синтез ДНК в репликатиной вилке, называют реплисомой. Белки, задействованные в репликативной вилке, перечислены в табл. 25-4.
Таблица 25-4. Белки реплисомы E. coli
|
Белок |
Мr |
Число субъединиц |
Функция |
|
SSB |
75 600 |
4 |
Связывание с одноцепочечной ДНК |
|
Белок DnaB (хеликаза) |
300 000 |
6 |
Раскручивание ДНК; компонент праймосомы |
|
Праймаза (белок DnaG) |
60 000 |
1 |
Синтез РНК-праймера; компонент праймосомы |
|
ДНК-полимераза III |
791 500 |
17 |
Элонгация новой цепи |
|
ДНК-полимераза I |
103 000 |
1 |
Заполнение брешей; вырезание праймеров |
|
ДНК-лигаза |
74 000 |
1 |
Лигирование |
|
ДНК-гираза (ДНК-топоизомераза II) |
400 000 |
4 |
Суперскручивание |
Рис. 25-14. Синтез ДНК на лидирующей и отстающей цепях. События, происходящие в репликативной вилке, координируются одним димером ДНК-полимеразы III в комплексе с DnaB-хеликазой. Здесь показан уже идущий процесс репликации (стадии от а до д обсуждаются в тексте). Отстающая цепь изогнута петлей, так что синтез ДНК происходит одновременно на лидирующей и на отстающей матрице. Красные стрелки указывают 3′-концы двух новых цепей и направление синтеза ДНК, толстые черные стрелки — направление движения родительской ДНК через комплекс. На отстающей цепи начинает синтезироваться фрагмент Оказаки. Цветовое обозначение субъединиц и функции погрузчика зажима объясняются на рис. 25-15.
Погрузчик зажима ДНК-полимеразы III. состоящий из частей двух Ʈ-субъединиц, а также из y, δ и δ’-субъединиц, функционирует, кроме того, как ААА+ АТРаза. Этот комплекс связывается с АТР и с новым скользящим β-зажимом. Связывание приводит к возникновению напряжения в димерном зажиме, в результате чего на границе одной из субъединиц открывается кольцо (рис. 25-15). Отстающая цепь с только что присоединенным праймером проскальзывает в кольцо через образующийся разрыв. Затем погрузчик зажима гидролизует АТР, высвобождая скользящий (3-зажим и позволяя ему сомкнуться вокруг ДНК.
Рис. 25-15. Комплекс погрузчика зажима ДНК-полимеразы III. Комплекс образован пятью субъединицами: субъединица y, δ и δ’, а также N-концевыми доменами обеих Ʈ-субъединиц (см. рис. 25-10). Комплекс связывает три молекулы АТР и димерный β-зажим. Это связывание приводит к открыванию β-зажима на границе одной из двух субъединиц. Гидролиз связанного АТР позволяет β-зажиму вновь замкнуться вокруг ДНК.
Реплисома осуществляет синтез ДНК быстро, присоединяя примерно по 1000 нуклеотидов в секунду к каждой цепи (лидирующей и отстающей). После завершения сборки одного фрагмента Оказаки его РНК-праймер удаляется и замещается последовательностью ДНК с помощью ДНК-полимеразы I, а оставшийся ник ликвидируется ДНК-лигазой (рис. 25-16).
Рис. 25-16. Заключительные стадии синтеза фрагментов отстающей цепи. ДНК-полимераза I благодаря своей 5′ —> 3′-экзонуклеазной активности удаляет РНК- праймеры из отстающей цепи и замещает их на ДНК. Оставшийся ник (разрыв) ликвидирует ДНК-лигаза. Роль АТР и NAD+ показана на рис. 25-17.
ДНК-лигаза катализирует образование фосфодиэфирной связи между 3’-гидроксильной группой на конце одной цепи ДНК и 5’-фосфатом на конце другой цепи. Фосфат должен быть предварительно активирован путем аденилирования. ДНК-лигазы, выделенные из вирусов и эукариот, используют для этой цели АТР. ДНК-лигазы бактерий (рис. 25-17) отличаются тем, что в качестве источника АМР они используют NAD+- кофактор, который обычно участвует в реакциях с переносом гидрид-иона (см. рис. 13-24). ДНК- лигаза еще один фермент метаболизма ДНК, который стал важным инструментом в экспериментах с рекомбинантными ДНК (см. рис. 9-1).
Рис. 25-17. Механизм реакции. Действие ДНК-лигазы. На каждой из трех стадий одна фосфодиэфирная связь образуется за счет другой. Стадии ① и ② приводят к активации 5′-фосфатной группы в нике. АМР сначала переносится на остаток лизина в молекуле фермента, а затем на 5′-фосфат в нике. На стадии ③ 3′-гидроксильная группа атакует этот фосфат и вытесняет АМР, образуя фосфодиэфирную связь и закрывая тем самым разрыв. У Е. coli АМР в реакции ДНК-лигазы происходит из NAD+. ДНК- лигазы, выделенные из некоторых вирусных и эукариотических источников, используют АТР, а не NAD+, и на стадии ① они высвобождают пирофосфат, а не никотина- мидмононуклеотид (NMN).
Терминация.
Наконец, две репликативные вилки кольцевой хромосомы Е. coli встречаются в точке терминации, содержащей множество копий последовательности из 20 п. н« называемой Теr (от лат. terminus — конец) (рис. 25-18). Последовательности Теr расположены на хромосоме таким образом, что создают что-то вроде ловушки, в которую репликативная вилка может войти, но не может из нее выйти. Теr-последовательности служат участками связывания белка Tus (от англ. terminus utilization substance). Комплекс Tus Ter может задержать репликативную вилку, движущуюся только в одном направлении. В каждом цикле репликации действует только один комплекс Tus- Теr — первый из тех, с которым столкнется первая вилка. Учитывая, что движущиеся в противоположных направлениях репликативные вилки обычно останавливаются при встрече, может показаться, что Теr-последовательности — необязательные элементы. Однако они могут предотвратить избыточную репликацию на одной из репликативных вилок, в случае если другая задерживается или останавливается при столкновении с повреждением ДНК или каким-либо другим препятствием.
Рис. 25-18. Терминация репликации хромосомы Е. coli. Последовательности Теr (от ТеrА до TerF) располагаются на хромосоме в виде двух кластеров с противоположной ориентацией.
Таким образом, когда одна из репликативных вилок встречает функциональный комплекс Tus-Ter, она останавливается; а другая вилка останавливается, когда встречается с первой (задержанной) вилкой. Затем реплицируются последние несколько сотен пар оснований ДНК между этими крупными белковыми комплексами (по неизвестному механизму), достраивая две топологически сцепленные (катенированные) кольцевые хромосомы (рис. 25-19). Кольцевые ДНК, связанные таким образом, называются катенанами. Функцию разделения катенированных колец у Е. coli выполняет топоизомераза IV (топоизомераза II типа). Затем при клеточном делении разделенные хромосомы расходятся по дочерним клеткам. Заключительная фаза репликации других кольцевых хромосом, включая многие из ДНК-содержащих вирусов, инфицирующих эукариотические клетки, происходит по аналогичной схеме.
Рис. 25-19. Роль топоизомераз в терминации репликации. В результате репликации ДНК на двух противоположно направленных репликативных вилках образуются полноценные хромосомы, соединенные как катанены (топологически соединенные кольца). Кольца не связаны ковалентной связью, но поскольку они переплетены и каждое из них замкнуто ковалентно, их нельзя разделить без помощи топоизомераз. У Е. coli основную роль в разделении катенированных хромосом играет топоизомераза типа II (ДНК-топоизомераза IV), которая вносит кратковременный разрыв в обе цепи ДНК в одной хромосоме, позволяя другой хромосоме пройти сквозь разрыв.
Репликация в эукариотических клетках происходит по похожей схеме, но сложнее
Молекулы ДНК в эукариотических клетках значительно крупнее, чем в бактериях, и образуют сложные нуклеопротеиновые структуры (хроматин; разд. 24.3). Основная схема репликации ДНК у эукариот такая же, как у бактерий, и многие белковые комплексы похожи по функциям и структуре. Однако репликация у эукариот регулируется и координируется в соответствии с клеточным циклом, что усложняет процесс.
Участки начала репликации достаточно хорошо охарактеризованы у некоторых низших эукариот, но значительно хуже у высших эукариот. У позвоночных для инициации репликации могут использоваться разные А = Т-богатые последовательности, причем участки начала репликации могут меняться при каждом клеточном делении. Репликация дрожжей Saccharomyces cerevisiae начинается в определенной области, называемой автономно реплицирующейся последовательностью (АРП) или репликатором. Дрожжевые репликаторы имеют длину примерно 150 п. н. и содержат несколько важных консервативных последовательностей. В 16 хромосомах гаплоидного генома дрожжей рассредоточено около 400 репликаторов.
Механизмы регуляции обеспечивают однократную репликацию всей клеточной ДНК в каждом клеточном цикле. Важную роль в этой регуляции играют белки, называемые циклинами, и циклин-зависимые киназы (CDK), с которыми они образуют комплексы (с. 660 в т. 1). Циклины быстро разрушаются в результате убиквитин- зависимого протеолиза в конце М-фазы (фазы митоза), а отсутствие циклинов способствует организации пререпликативных комплексов (рrе- RC) в участках инициации репликации. В быстро растущих клетках комплекс pre-RC образуется в конце М-фазы. В медленно растущих клетках этот комплекс не образуется до конца фазы G1. Формирование комплексов pre-RC позволяет клетке начать репликацию (этот этап иногда называют лицензированием).
Как и у бактерий, ключевой момент в инициации репликации у всех эукариот — присоединение репликативной хеликазы, гетерогексамерного белкового комплекса МСМ (от англ. minichromosome maintenance; субъединицы от МСМ2 до МСМ7). Кольцевая хеликаза, состоящая из субъединиц МСМ2-7, функционирует подобно бактериальной хеликазе DnaB и связывается с ДНК при помощи другого комплекса из шести белков, называемого комплексом распознавания точки инициации репликации (ORC; от англ. origin recognition complex; рис. 25-20). Комплекс ORC состоит из пяти ААА+ АТРазных доменов и по своим функциям напоминает бактериальный комплекс DnaA. Кроме того, для присоединения комплекса МСМ2-7 необходимы два белка — CDC6 (от англ. cell division cycle) и CDT1 (от англ. CDC 10-dependent transcript 1), причем дрожжевой белок CDC6 представляет собой еще одну ААА+ АТРазу.
Рис. 25-20. Сборка пререпликативного комплекса в точке начала репликации у эукариот. В участке инициации репликации связываются белки ORC, CDC6 и CDT1. Эти белки, многие из которых являются ААА+ АТРазами, способствуют присоединению репликационной хеликазы МСМ2-7 в реакции, аналогичной реакции присоединения бактериальной хеликазы DnaB с помощью белка DnaC. В результате связывания хеликазного комплекса МСМ с ДНК образуется пререпликативный комплекс pre-RC, и именно эта стадия оказывается ключевой для инициации репликации.
Для осуществления репликации в S-фазе происходит синтез и активация комплексов циклин-CDK (таких, как комплекс циклин Е- CDK2; см. рис. 12-45 в т. 1) и CDC7-DBF4. Оба типа комплексов помогают активизировать репликацию путем связывания и фосфорилирования некоторых белков пререпликативных комплексов. Другие циклины и CDK ингибируют образование дополнительных pre-RC комплексов после начала репликации. Например, CDK2 связывается с циклином А, когда уровень циклина Е снижается в ходе S-фазы, что ингибирует активность CDK2 и предотвращает образование дополнительных pre-RC-комплексов.
Скорость репликативной вилки у эукариот (около 50 нуклеотидов в секунду) примерно в 20 раз меньше, чем в Е. coli. Если бы репликация хромосомы человека начиналась в единственной точке инициации репликации, при такой скорости процесса репликация средней хромосомы продолжалась бы более 500 ч. На самом деле репликация хромосом человека происходит в двух направлениях и начинается во многих точках, удаленных друг от друга на расстояние от 30 до 300 т. п. н. Хромосомы эукариот почти всегда намного длиннее, чем хромосомы бактерий, поэтому наличие многих точек инициации репликации, вероятно, общее свойство клеток эукариот.
Как и у бактерий, у эукариот есть несколько типов ДНК-полимераз. Некоторые из них, возможно, выполняют особые функции, например, репликацию митохондриальной ДНК. В репликации ядерных хромосом участвует ДНК-полимераза а в комплексе с ДНК-полимеразой δ. Субъединичная ДНК-полимераза α имеет похожую структуру и свойства во всех эукариотических клетках. Одна из ее субъединиц выполняет функцию праймазы, а самая крупная субъединица (Мr ≈ 180 000) обладает полимеразной активностью. Однако данная полимераза не имеет корректирующей 3′ —> 5′-экзонуклеазной активности и не может обеспечить высокой точности репликации. Предполагается, что ДНК-полимераза α нужна только для синтеза коротких праймеров (либо РНК, либо ДНК) для фрагментов Оказаки на отстающей цепи. Удлинение этих праймеров осуществляет мультисубъединичная ДНК -полимераза δ. Этот фермент связывается с белком, называемым ядерным антигеном пролиферирующих клеток (PCNA — от англ. proliferating cell nuclear antigen; Mr = 29 000), и стимулируется им; этот белок в большом количестве содержится в ядрах делящихся клеток. Трехмерная структура PCNA удивительным образом напоминает структуру β-субъединицы ДНК-полимеразы III Е. соli (рис. 25-10, б), хотя первичные последовательности этих белков не имеют выраженной гомологии. Функционально PCNA представляет собой аналог β-субъединиц; он формирует кольцевой зажим, который существенно увеличивает процессивность полимеразы. ДНК-полимераза δ обладает корректирующей 3′ —> 5′-экзонуклеазной активностью и, предположительно, обеспечивает синтез как лидирующей, так и отстающей цепи в комплексе, напоминающем димерную бактериальную ДНК-полимеразу III.
Еще одна полимераза, ДНК-полимераза ε, в некоторых ситуациях заменяет ДНК-полимеразу δ, например, при репарации ДНК. ДНК-полимераза е также может действовать в репликативной вилке, возможно, аналогично бактериальной ДНК-полимеразе I, удаляя праймеры фрагментов Оказаки на отстающей цепи.
В репликации ДНК у эукариот задействованы еще два белковых комплекса. Белок RPA (от англ. replication protein А) связывает одноцепочечную ДНК, и его функция аналогична функции белка SSB в клетках Е. coli Белок RFC (от англ/ replication factor С) представляет собой погрузчик зажима для PCNA, который облегчает сборку активных репликационных комплексов. Аминокислотные последовательности субъединиц комплекса RFC имеют значительное сходство с последовательностями субъединиц бактериального y-комплекса.
Для терминации репликации линейных эукариотических хромосом на концах каждой хромосомы синтезируются специальные структуры, называемые теломерами (см. гл. 26).
Вирусные ДНК-полимеразы являются мишенями для противовирусной терапии
Многие ДНК-co держащие вирусы кодируют собственные ДНК-полимеразы, и некоторые из них становятся мишенями для лекарственных средств. Например, ДНК-полимераза вируса простого герпеса ингибируется ацикловиром, разработанным Гертрудой Элайон (см. с. 558 в т. 2). Ацикловир состоит из гуанина, к которому присоединено неполное кольцо рибозы.
Ацикловир фосфорилируется вирусной тимидинкиназой. Сродство ацикловира к вирусному ферменту в 200 раз выше сродства к клеточной тимидинкиназе. В результате фосфорилирование происходит преимущественно в клетках, инфицированных вирусом. Клеточные киназы превращают образовавшийся ацикло-GMP в ацикло-GTP, который одновременно является и ингибитором, и субстратом ДНК-полимераз. Ацикло-GTP сильнее ингибирует ДНК-полимеразу вируса герпеса, чем клеточные ДНК-полимеразы. Поскольку ацикло-GTP не имеет 3′-гидроксильной группы, его включение в цепь ДНК воспринимается как сигнал терминации. Таким образом, репликация вируса ингибируется на нескольких стадиях. ■
Краткое содержание раздела 25.1 Репликация ДНК
■ Репликация ДНК происходит с очень высокой точностью и в определенной фазе клеточного цикла. Репликация полуконсервативна, каждая цепь выступает матрицей для новой дочерней цепи. Репликация протекает в три стадии: инициация, элонгация и терминация. В бактериях реакция начинается в точке инициации репликации и обычно происходит в двух направлениях.
■ ДНК синтезируется ДНК-полимеразами в направлении 5′ —> 3′. В репликативной вилке лидирующая цепь синтезируется непрерывно по ходу движения репликативной вилки; отстающая цепь синтезируется прерывисто в виде фрагментов Оказаки, которые затем сшиваются.
■ Точность репликации ДНК достигается (1) путем выбора оснований полимеразой, (2) с помощью корректирующей 3′ —> 5′-экзонуклеазной активности, которой обладает большинство ДНК-полимераз, и (3) с помощью специфических систем репарации, исправляющими ошибки, оставшиеся после репликации.
■ Большинство клеток имеет несколько ДНК-полимераз. У Е. coli основной фермент репликации — ДНК-полимераза III. ДНК-полимераза I выполняет особые функции в ходе репликации, рекомбинации и репарации.
■ На стадиях инициации, элонгации и терминации репликации ДНК работает множество ферментов и белковых факторов, многие из них принадлежат к семейству ААА+ АТРаз.
■ Белки бактерий, участвующие в репликации, организованы в крупные комплексы, внутри которых матрица ДНК протягивается через две реплисомы, связанные с плазматической мембраной.
рис.1 Модифицированные азотистые основания ДНК, удаляемые ДНК–гликозилазами: а – урацил; б – гипоксантин; в – 5–гидроксицитозин; г – 2,5-диамино-4-формамидопиримидин; д – 7,8-дигидро-8-оксогуанин; е – мочевина; ж – тимингликоль; з – 5-формилурацил; и – 5-гидроксиметилурацил; к – 3-метиладенин; л – 7-метилгуанин; м – 2-метилцитозин [Партушев, 2000].
АP-эндонуклеаза создает ник (одноцепочечный разрав) с 3’-ОН и 5’-dRP концами. Для большинства млекопитающих характерен
тип BER репарации с включением одного нуклеотида. В этом случае ДНК-полимераза β вставляет 1 нуклеотид в 3’-конец праймера и затем удаляет 5’-dRP фрагмент с помощью своей dRP-лиазной активности. Получившийся ник сшивается ДНК-лигазой. Альтернативным является путь так называемой длинно-заплаточной BER репарации. Он реализуется в тех случаях, когда 5’-dRP фрагмент модифицирован и не может быть удален с помощью ДНК–полимеразы β. В этом случае Pol β проводит синтез ДНК с вытеснением запирающей цепи, 2 — 13 нуклеотидов. Образующийся в результате свисающий 5’-конец ДНК выщепляется флэпэндонуклеазой FEN1. Pol бета, Pol дельтаи Pol епсилон могут вести ситнез ДНК во время длинно-заплаточной репарации. Идентичность полимераз, вовлеченных в этот процесс in vitro, еще не ясна, но показано, что Pol β всегда инициирует синтез ДНК.
Зависимость того, какой из путей BER реализуется, определяется бифункциональными ДНК-гликозилазами, которые имеют дополнительную
АP-лиазную активность. При комбинации ДНК-гликозилазной, АP-лиазной и АP-эндонуклеазной активностей внутри одного фермента в поврежденной цепи ДНК образуется однонуклеотидная брешь с 3’-ОН и 5’-P концами, которую может застроить Pol β.
Дополнительная сложность в понимании механизмов переключения внутри BER состоит в том, что две новые полимеразы Pol i и Pol лямбда также имеют dRP-лиазнуюактивность. Таким образом, можно предположить, что они, как и Pol бета, являются участниками тех процессов репарации, где необходимо выщепление dRP фрагмента. Pol лямбда является близким гомологом Pol бета со сходными ферментативными свойствами. Также как и Pol бета, она лишена 3’—>5’экзонуклеазной активности и имеет низкую процессивность синтеза на частичном ДНК-дуплексе, содержащем свисающий 5’-участок матрицы, процессивный синтез возможен в брешах с 5’-P концами. Таким образом, Pol лямбда является подходящим кандидатом для BER синтеза. Более того,
Pol лямбда заменяет Pol бета в реконструированных BER системах, репарирующих урацил-содержащие ДНК, iv vitro. Однако, клетки мышей Pol лямбда -/- не чувствительны к обработке перекисью или метилметансульфонатом, агентам, которые, помимо других типов повреждений, продуцируют АР-сайты и окисленные формы оснований. Отсюда можно заключить, что Pol лямбда не является необходимой для BER в клетках, где есть, Pol дельта и Pol епсилон. Интересно, что Pol лямбда может эффективно процессировать ДНК при очень низкой концентрации dNTP (около 1 мкМ), что может означать ее участие в любых клеточных процессах в фазе G0 при низкой концентрации dNTP. В поддержку этой гипотезы выступает тот факт, что экспрессия Pol лямбда зависит от клеточного цикла, и наибольшее количества белка экспрессируется при переходе из S- в М-фазу и в спокойных клетках.
Pol i принадлежит к Y семейству полимераз. Основная функция полимераз этого семейства – утилизация ДНК-повреждений, блокирующих работу репликативных ДНК-полимераз. Однако,
исходя из некоторых свойств Pol i, можно предположить, что этот фермент является участником альтернативного процесса BER репарации. Pol i обладает низкой поцессивностью, лишена 3’>5’экзонуклеазной активности и способна застраивать брешь в 1 — 5 нуклеотидов iv vitro. Pol i может замещать Pol бета в реконструированных системах BER, репарирующих урацил-содержащие ДНК, iv vitro, благодаря наличию dRP-лиазной и ДНК-полимеразной активностей. Кинетические исследования реакции нуклеотидного встраивания овыявили дополнительные данные, свидетельствующие в пользу того, что Pol i может принимать участие в репарации. Pol i вставляет dTMP напротив А в ДНК-матрицу с большей эффективностью, чем любой другой дезоксинуклеозидмонофосфат, при этом точность ДНК-синтеза сопоставима с параметрами для Pol бета. Основываясь на этих данных, можно предположить, что Pol i – участник BER репарации оснований уридина, который образуется после встраивания dUMP напротив А во время ошибочной репликации. Pol i также
может вставлять dGМP напротив Т со скоростями, близкими к скорости включения корректного нуклеотида. Более того, на матрице, содержащей 2 или более последовательных Т, вторым встраиваемым нуклеотидом оказался dGMP. Подобные результаты служат основой для гипотезы, что Pol i может быть участником альтернативной BER репарации в тех слсучаях, когда dG был ошибочно удален гликозилазой из G-T или G-U некомплементарной пары, образовавшейся в результате дезаминации 5-метилцитозина или цитозина. Возможная роль Pol i в трансляции синтеза ДНК и процессе соматических гипермутаций рассмотрена ниже.
Возможность участия пяти ядерных полимераз в BER, три из которых обладают dRP-лиазной активностью, была мало изучена в клетках и на модельных животных, лишенных более чем одной полимеразы. Поэтому данные о субстратной специфичности и взаимодействии полимеразо-акцепторных белков, необходимых в BER, недостаточны. Помимо BER репарации, Pol бетта принимает участие в репарации одноцепочечных разрывов; функционирование
этого процесса нарушено у пациентов с наследственной спинномозговой атаксией. Одноцепочечные разрывы генерируются эндогенными или экзогенными агентами. Такие разрывы часто содержат однонуклеотидные бреши с 3’ и/или 5’ модифицированными концами, то есть очень похожи на субстраты BER. Так вот, интересно посмотреть, способны ли другие полимеразы с подобным набором исходных данных (Pol i и Pol лямбда) принимать участие в репарации одноцепочечных разрывов.
Четвертой ДНК-полимеразой в клетках человека, обладающей dRP-лиазной активностью, является митохондриальная ДНК-полимераза гамма. Pol гамма – единственная ДНК-полимераза, обнаруженная в митохондриях, следовательно, она ответственна за все преобразованиях ДНК, происходящие в этой органелле. Митохондрия является объектом интенсивного повреждения ДНК активными формами кислорода, генерируемыми во время окислительного фосфорилирования. Эти повреждения эффективно репарируются набором митохондриальных белков, которые включают в себя АP-эндонуклеазу, Pol гамма,
мтДНК-лигазу. Сам процесс репарации сходен с однонуклеотидным процессом BER в ядерной ДНК.
Таблица. ДНК-гликозилазы и эндонуклеазы клеток микроорганизмов и человека, участвующие в BER.
В универсальном механизме эксцизионной репарации как прокариоты, так и эукариоты гидролизуют 3–5-ю фосфодиэфирную связь с 3′-конца отповреждения. При этом прокариоты гидролизуют также 8-ю связьот 5’-конца измененного нуклеотида, тогда как у эукариотических организмовпроисходит одноцепочечный разрыв на расстоянии 21–25 нуклеотидов отповреждения со стороны его 5’-конца. Таким образом, прокариоты удаляютизмененный нуклеотид в составе 12–13-членных олигомеров, тогда как
эукариоты – в составе одноцепочечных фрагментов ДНК длиной в 27–29 нуклеотидов. Ферментная система, вносящая такие двойные одноцепочечные
разрывы, получила название эксцизионной нуклеазы (эксцинуклеазы). Образующаяся в молекуле репарируемой ДНК одноцепочечная брешь далее заполняется с помощью ДНК-полимеразы, а фосфодиэфирная связь в остающемся одноцепочечном разрыве восстанавливается ДНК-лигазой.
NER устраняет ДНК-повреждения посредством вырезания олигонуклеотида, содержащего это повреждение, с образованием бреши в ДНК размером ~ 30 нуклеотидов. Эта брешь застраивается одной или двумя полимеразами В семейства Pol дельта или Pol епсилон. Любая из этих ДНК-полимераз способна застраивать подобную брешь в реконструированной системе, содержащей очищенные белки млекопитающих, in vitro. При изучении NER в системе фибробластов человека с нарушенной проницаемостью и в ядерном экстракте HeLa клеток обнаружили, что обе полимеразы необходимы для восстановления ДНК после УФ-облучения. Исследования
в дрожжевых системах показали, что та или другая ДНК-полимераза существенно дополняют репарацию УФ-поврежденной ДНК. Более поздние исследования показали, что оба фермента необходимы для эффективной NER репарации в дрожжевых экстрактах. Pol дельта и Pol епсилон способны вести процессивный синтез ДНК при наличии дополнительного фактора PCNA, который «надевается» на ДНК с помощью пятисубъединичного комплекса RFC. Репарационный синтез как Pol дельта, так и Pol епсилон также требует присутствия этих факторов процессивности.
Эксцизионная репарация нуклеотидов: введение
ЭР — эксцизионная репарация ДНК; ЭР-комплекс включает ПАРП , XRCC1 , ДНК-лигазу III и ДНК-полимеразу бета .
Два пути осуществления NER:
1. Гидролиз фосфодиэфирной связи по 3′- или 5′- концу на некотором расстоянии от ошибочно спаренного (поврежденного) нуклеотида, который далее целиком удаляется под действием 5′->3′- (или 3′->5′-) экзонуклеазы, гидролизующей
цепь ДНК нуклеотид за нуклеотидом в соответствующем направлении от первоначального одноцепочечного разрыва в репарируемой ДНК. Образующаяся брешь далее заполняется ДНК-полимеразой. Такой механизм репарации реализуется у E. coli и человека для вырезания неповрежденных (немодифицированных) ошибочно спаренных нуклеотидов. Механизм последовательного эндо- и экзонуклеазного расщепления ДНК не используется для удаления поврежденных (измененных) нуклеотидов. Это связано с тем, что такие нуклеотиды часто являются ингибиторами экзонуклеаз.
2. Функционирует у всех исследованных видов организмов и заключается в использовании ферментной системы, которая вносит одноцепочечные разрывы по обе стороны от поврежденного нуклеотида на некотором расстоянии от него с последующим удалением одноцепочечного фрагмента ДНК, содержащего измененный нуклеотид.
Гидролизуется 3-5-фосфодиэфирную связь с 3′-конца от повреждения. При этом прокариоты гидролизуют также 8-связь от 5′-конца
измененного нуклеотида, тогда как у эукариотических организмов происходит одноцепочечный разрыв на расстоянии 21-25 нуклеотидов от повреждения со стороны его 5′-конца. Таким образом, прокариоты удаляют измененный нуклеотид в составе 12-13-членных олигомеров, тогда как эукариоты — в составе одноцепочечных фрагментов ДНК длиной в 27-29 нуклеотидов. Ферментная система, вносящая такие двойные одноцепочечные разрывы, получила название эксцизионной нуклеазы (эксцинуклеазы) . Образующаяся в молекуле репарируемой ДНК одноцепочечная брешь далее заполняется с помощью ДНК-полимеразы, а фосфодиэфирная связь в остающемся одноцепочечном разрыве восстанавливается ДНК-лигазой.
В отличие от ЭРО и прямых реверсий, которые специфичны к достаточно узкому кругу повреждений ДНК, система ЭРН, хотя и с разной эффективностью, удаляет все возможные повреждения, и потому роль ее в поддержании стабильности генома велика. ЭРН детально изучена у E. coli и активно изучается в клетках
дрожжей и человека, причем в последних благодаря раскрытию генетической природы таких заболеваний, как пигментная ксеродерма ( ХР ), синдром Кокейна ( СS ) и заболевания трихотиодистрофия ( ТТD)
В ЭРН задействовано 6-8 генов у E. coli и до 30 у человека.
Если повреждение в транскрибируемой (матричной) нити ДНК задерживает продвижение РНК-полимеразы, то специальный белковый фактор TRCF ( transcription-repair coupling factor ) сталкивает РНК-полимеразу и связывает с этим местом комплекс ферментов репарации. См. Сопряжение транскрипции и репарации ДНК у E/coli.
Ферментный ансамбль осуществляющий первые 3 стадии процесса NER называется эксцинуклеазой.
В системе эксцизионной репарации ДНК путем удаления нуклеотидов поврежденные азотистые основания вырезаются в составе олигонуклеотидов.
NER: регуляция
Для клеток животных не характерен SOS-ответ, свойственный клеткам E. coli и представляющий собой суммарную реакцию бактериальной клетки на повреждение ДНК различными агентами,
проявляющийся в усилении транскрипции генов NER. Посттрансляционные модификации белков репарации, происходящие в ответ на повреждение ДНК, не влияют на активность эксцинуклеазы человека.
Обнаружено, что повреждения ДНК стабилизирует белок р53- белок-супрессор опухолевого роста, являющийся регулятором транскрипции. Имеются данные о том, что белок р53 может взаимодействовать с белками XPB и RPA, необходимыми для NER. Однако клетки с инактивированными генами р53 (p53(-/-)), как и клетки дикого типа, эффективно удаляют из поврежденной ДНК два основных фотопродукта, возникающих под действием УФ-света, и обладают такой же устойчивостью к УФ. Поэтому считается, что белок р53 не оказывает прямого влияния на NER. Белки Cdk7 и циклин H, которые образуют Cdk-активирующую киназу, входят в состав комплекса TFIIH, что позволяет предполагать наличие связи репарации ДНК с фазами клеточного цикла.
ЭРН (NER): сопряжение с транскрипцией (ТЭРН)
Транскрибируемые последовательности нуклеотидов ДНК, особенно
в матричной цепи, репарируются с большей скоростью, чем нетранскрибируемые последовательности. В клетках больных с синдромом Кокайна не наблюдается такой асимметрии в репарации.
В клетках E. coli белковый фактор, кодируемый геном mfd и сопрягающий транскрипцию и репарацию, замещает остановившиеся перед повреждением молекулы РНК-полимеразы, что приводит к диссоциации транскрипционного комплекса. При этом он привлекает экзонуклеазный репаративный комплекс к поврежденному участку ДНК. См. Сопряжение транскрипции и репарации ДНК (ТЭРН) у E/coli
В клетках животных ген CSB кодирует белок с молекулярной массой 160 кДа, который содержит хеликазный домен и, возможно, выполняет те же функции, что и белок Mfd у E. coli . На основе поведения клеток с мутантными генами белков CSA и CSB разработана модель механизма, с помощью которого обеспечивается асимметричная репарация цепей ДНК. В соответствии с этой моделью РНК-полимераза II , остановившаяся в процессе транскрипции
перед поврежденным участком ДНК, распознается комплексом CSA-CSB и перемещается в сторону от повреждения без разрушения четвертичной структуры транскрипционного комплекса. Одновременно комплекс CSA-CSB привлекает компоненты репаративной системы XPA и TFIIH к месту повреждения ДНК и помогает сборке эксцинуклеазного комплекса. Нуклеотиды поврежденной цепи вырезаются, и брешь репарируется. После этого РНК-полимераза в составе транскрипционного комплекса продолжает транскрипцию.
NER: механизм
Процесс NER можно разделить на четыре этапа: а) распознавание поврежденного участка ДНК; б) двойное надрезание (инцизия) цепи ДНК по обеим сторонам поврежденного участка и его удаление (эксцизия); в) заполнение бреши в процессе репаративного синтеза; г) лигирование оставшегося одноцепочечного разрыва ДНК.
NER человека распознает и удаляет одиночные ошибочно спаренные нуклеотиды, а также петли длиной в 1-3 нуклеотида. NER человека способна различать цепи ДНК в случае распознавания поврежденных
нуклеотидов. Показано, что при наличии в ДНК димеров тимина циклобутанового типа вырезание нуклеотидов происходит исключительно из поврежденной цепи. Механизм такого распознавания в настоящее время неизвестен, как и молекулярный механизм узнавания самих поврежденных оснований. Система способна распознавать повреждения как сильно, так и слабо деформирующие вторичную структуру ДНК. При этом не обнаружена линейная зависимость между коэффициентом специфичности нуклеазы (kcat/km) и уровнем деформации двойной спирали ДНК. Показано, что в процессе распознавания участвуют белковые комплексы XPA/RPA, которые преимущественно связываются с поврежденной ДНК, и TFIIH, обладающий АTP-зависимой ДНК-расплетающей активностью. Последний взаимодействует с поврежденным участком ДНК и по аналогии с соответствующим механизмом у E. coli локально раскручивает ДНК, создавая основной преинцизионный комплекс с поврежденной ДНК.
Установлено, что три фермента репарации, обладающие узкой
субстратной специфичностью: ДНК-фотолиаза (удаление пиримидиновых димеров), урацилгликозилаза (удаление урацила из ДНК) и экзонуклеаза III (гидролиз ДНК в AP-сайтах), втягивают поврежденный участок из двойной спирали в полость фермента, что приводит кофактор или аминокислотные остатки активного центра этих ферментов в непосредственный контакт с расщепляемыми связями ДНК. Не исключено, что система эксцинуклеазы действует таким же образом.
Основные этапы функционирования NER, следующие за распознаванием поврежденного участка ДНК, представлены на рис. I.59. После связывания комплекса XPA-RPA с измененным участком ДНК, XPA взаимодействует с комплексом TFIIH, который создает преинцизионный комплекс, что сопряжено с гидролизом ATP. ATP-зависимое расплетание ДНК комплексом TFIIH подготавливает ее к взаимодействию с двумя XP-белками, обладающими нуклеазной активностью. XPG связывается с TFIIH и вносит одноцепочечный разрыв с 3′-конца повреждения. Аналогично комплекс ERCC1-XPF
взаимодействует с XPA в составе преинцизионного комплекса и способствует одноцепочечному разрыву с 5′-конца повреждения. Образование обоих разрывов является ATP-зависимым, и их расположение на ДНК высокоспецифично. Как правило, происходят разрывы 5-й и 24-й фосфодиэфирных связей соответственно от 3′- и 5′-концов поврежденных участков. Однако расположение точек разрывов может варьироваться.
Таким образом, в результате подобных одноцепочечных надрезов ДНК может освобождаться фрагмент длиной 24-32 нуклеотида с преобладанием фрагментов длиной 27-29 нуклеотидов. На расположение сайтов одноцепочечных разрывов влияют характер повреждения и последовательности нуклеотидов (контекст), окружающих поврежденный участок. Ту же самую картину инцизии обнаруживают in vivo в ооцитах Xenopus и у Schizosaccharomyces pombe. На этом основании делают вывод об универсальном механизме эксцизионной репарации у эукариот.
Репаративный синтез ДНК у человека является PCNA-зависимым,
т.е. может осуществляться с участием ДНК-полимераз Polдельта и Polэпсилон. PCNA связывается с системой праймер-матрица под действием фактора репликации RFC, откуда следует, что последний также участвует в репаративном синтезе ДНК. В опытах с бесклеточными системами моноклональные антитела к Polдельта специфически подавляют репаративный синтез. Однако оказалось, что в тех же высокоочищенных бесклеточных системах вместо Polдельта с аналогичным эффектом могут быть использованы Polэпсилон и даже фрагмент Кленова ДНК-полимеразы I E. coli. Это означает, что реконструированные из очищенных компонентов бесклеточные системы лишь в ограниченной степени имитируют биохимические процессы, происходящие в живых клетках. Считается, что обе ДНК-полимеразы — Polдельта и Polэпсилон участвуют в репаративном синтезе ДНК у человека.
ПАРП: модели управления процессом эксцизионной репарации ДНК
ПАРП — один из первых ядерных факторов, распознающих повреждение ДНК , и поэтому в идеальном случае управляет запуском механизма
репарации ДНК в живых клетках с места повреждения ДНК. Эта модель поддерживается идентификацией ЭР -комплекса, включающего ПАРП , XRCC1 , ДНК-лигазу III и ДНК-полимеразу бета . Присутствие ПАРП в таком мультипротеиновом комплексе доказывает, что этот фермент может направлять аппарат репарации ДНК к сайтам повреждения ДНК in vivo и облегчает осуществление репарации по этому пути.
XRCC1-белок действует как молекулярные «леса», формируя ЭР-комплекс путем индивидуального взаимодействия с каждым компонентом. С тех пор, как была обнаружена возможность поли(АДФ-рибозил)ирования XRCC1-белка in vitro, можно предположить, что ПАРП способен регулировать активность комплекса путем модифицирования XRCC1-белка in vivo и нарушать его способность взаимодействовать с другими компонентами комплекса. Было показано, что сверхэкспрессия XRCC1-белка подавляет активность ПАРП в живых клетках.
Аналогично ДНК-лигаза III ингибирует активность ПАРП in vitro, когда ее количества превышают количества ПАРП.
ПАРП может также рекрутировать факторы репарации ДНК путем модификации хроматиновых белков. Длинные цепи ПАР действительно способны направить ферменты репарации к сайтам разрывов ДНК значительно быстрее, нежели если они ищут повреждение сами по всему ядру. Такая модель согласуется с активностью ПАРП перед и/или после удаления поврежденных оснований.
NER: структура и функции белков
В табл. I.21 суммированы свойства белков животных, участвующих в NER. Большинство этих белков существует in vivo в виде комплексов, поэтому ферментативные активности, обнаруживаемые у отдельных белков в очищенном состоянии, могут не иметь прямого отношения к их функциям в системе NER.
XPA — белок с молекулярной массой 31 кДа, обладает доменом типа «цинковые пальцы», участвует в распознавании поврежденного участка ДНК, взаимодействует с другими компонентами системы и может функционировать в качестве фактора нуклеации для экзонуклеазы. XPA взаимодействует своим N-концевым доменом
с гетеродимером ERCC1-XPF, а С-концевым доменом — с TFIIH.
Белок RPA (HSSB) образует комплекс с XPA и усиливает его специфичность в отношении поврежденной ДНК. RPA (HSSB) — тример, состоящий из белковых субъединиц р70, р34 и р11, необходим для репликации ДНК и репаративного синтеза, а также для прохождения этапа двойного надреза ДНК во время эксцизионной репарации. Он обладает умеренным сродством к поврежденной ДНК.
TFIIH — олигомерный комплекс, в состав которого входят белки р89, р80, р62, р44, р41, р38 и р34. Этот белковый комплекс был открыт как один из семи основных факторов транскрипции, необходимых для эффективного функционирования РНК-полимеразы II . Субъединица р89 идентична белку репаративного комплекса XPB. Обнаружено отсутствие функциональной комплементации между бесклеточными экстрактами клеток с мутантными белками XPB и XPD, определяемой по восстановлению репарирующей активности в смешанных экстрактах. Комплекс TFIIH представляет собой
фактор репаративной системы. Белки XPB и XPD являются ДНК-зависимыми АТРазами, обладают хеликазными доменами и могут (как и сам фактор TFIIH) вызывать диссоциацию гибридов, образованных между короткими фрагментами ДНК и одноцепочечной ДНК.
XPC — белок с молекулярной массой около 125 кДа, существует в виде гетеродимера в комплексе с белком р58, который является гомологом белка Rad23 дрожжей (HHR23B). XPC слабо связывается с TFIIH и очень прочно — с одноцепочечной ДНК.
ERCC1/XPF — прочный белковый комплекс, с которым взаимодействует белок XPA, обладающий эндонуклеазной активностью, специфичной в отношении одноцепочечной ДНК.
XPG — белковый комплекс, обладающий эндонуклеазной активностью, специфичной в отношении одноцепочечной ДНК; вовлекается в эксцизионный комплекс посредством взаимодействия с TFIIH и RPA.
ЭРН (NER): генетика
Гены NER E. coli, uvrA, uvrB и uvrC не обнаруживают гомологии с соответствующими генами человека. Гены NER дрожжей и человека
высокогомологичны, и энзимология эксцизионной репарации также обладает большим сходством. По крайней мере, три заболевания у человека вызываются генетическими нарушениями системы эксцизионной репарации: пигментная ксеродерма, синдром Кокейна и трихотиодистрофия.
Кожа больных пигментной ксеродермой обладает повышенной чувствительностью к дневному свету, что проявляется в виде фотодерматозов, включая рак кожи. В ряде случаев отмечены аномалии нервной системы, причиной которых являются мутации в одном из семи генов: XPA, XPB, XPC, XPD, XPE, XPF, XPG. Однако описаны больные с классическими симптомами пигментной ксеродермы, но с ненарушенной системой NER. Для клеток этих больных характерны изменения в так называемой пострепликативной репарации .
Больным с синдромом Кокейна присущи нарушения роста, умственная отсталость, катаракты, повышенная чувствительность к свету с сопутствующими дерматозами. Обнаружены мутации в двух группах генов, приводящие к этому заболеванию. У больных
с мутантными генами CS-A или CS-B клетки способны нормально репарировать УФ-повреждения ДНК. У другой группы больных обнаружены мутации в генах XPB, XPD или XPG.
У больных трихотиодистрофией со смешанными симптомами выявлены мутации в генах XPB или XPD. Классические симптомы этого заболевания, по-видимому, являются следствием мутации в гене транскрипционного фактора TFIIH.
Получение мутантов с измененной NER у грызунов позволило разбить такие гены на 11 групп комплементации, большинство из которых соответствует группам комплементации XP и CS человека. Часть соответствующих генов человека удалось клонировать, используя их способность исправлять (комплементировать) генетические дефекты в культивируемых мутантных клетках грызунов. Эти гены получили название кросс-комплементирующих генов эксцизионной репарации ( ERCC — excision repair cross complementing ). Среди них гены XPE и ERCC6-ERCC11 не требовались для прохождения основных реакций эксцизионной репарации, и их функция неизвестна.
Эксцизионная репарация нуклеотидов (ЭРН) E. coli
У E. coli эксцинуклеаза формируется, в результате димеризации двух молекул белка UvrА в присутствии АТР и связывания с одной молекулой UvrB. Гетеротример UvrA2B связывается с ДНК и с помощью 5′-3′-ДНК-геликазной активности перемещается вдоль ДНК в поисках повреждения. Такова предполагаемая картина узнавания эксцинуклеазой UvrAВС неспецифического повреждения в ДНК [ Hoeijmakers J., 1993 ]. Результатом узнавания повреждения этим комплексом явится внедрение субъединицы UvrB в ДНК, сопровождающееся конформационными изменениями ДНК в сайте внедрения ( изломы , локальная денатурация), диссоциация обеих субъединиц UvrА из комплекса и последующее связывание с ним молекул UvrD и UvrC. Гетеродимер UvrBC из состава нового комплекса делает два разрыва в поврежденной нити ДНК на расстоянии 8 н. с 5′-конца от повреждения и 5 н. с 3′-конца, катализируемых субъединицами UvrC и UvrB соответственно, что приводит к появлению 12-13-мерного олигонуклеотида,
вытесняемого из комплекса с помощью геликазы UvrD. Образующаяся при этом брешь заполняется ДНК-полимеразой I, синтезирующей ДНК по неповрежденной матрице, и сшивается ДНК-лигазой. Описанный процесс зависит от АТР. Действительно, АТР стимулирует димеризацию молекул UvrA, гидролиз АТР необходим для образования комплекса UvrA2B и проявления его геликазной активности, АТР необходим для формирования комплекса UvrB-ДНК и, наконец, для описанной выше бимодальной инцизии ДНК [ Friedberg E.C., Walker G.C., ea., 1991 ]. У E. coli транскрипционно-зависимую ветвь ЭРН (ТЭРН) осуществляет продукт гена mfd (mutation frequency decline). Этот ген был открыт за 35 лет до того, как его продукт признали фактором TRCF. Последний способствует диссоциации РНК-полимеразы от дефектной транскрибируемой нити ДНК, и связывается с субъединицей UvrA эксцинуклеазного комплекса. Иными словами, именно белок Mfd отыскивает повреждения на транскрибируемой нити и направляет ТЭРН на эту мишень.
Согласно недавним наблюдениям, два главных белка системы ДКНО — MutL и MutS — необходимы для ТЭРН [106 Melon I., Champl G.N., 1996106]. Причина такой взаимосвязи систем ДКНО и ТЭРН или заимствования белков MutL и MutS для выполнения сходных функций пока остается загадочной, хотя сам факт несомненен и нашел подтверждение также и для белков Msh2, Mlh1, Pms1 и Msh3 у S. cerevisiae [ Sweder K.S., ea, 1996 ].
Эксцизионная репарация нуклеотидов (ЭРН) у человека
Большому прогрессу в раскрытии механизма ЭРН у эукариот мы обязаны наследственному заболеванию человека — пигментной ксеродерме .
Изучение молекулярных основ этого заболевания выявили их сопряженность с дефектами системы ЭРН, результатом чего и явилось раскрытие генетического контроля ЭРН. Комплементационный анализ различных клеточных линий ХР определил 8 комплементационных групп (7 от XPA до XPG и одна «вариантная форма» XPV) [ Friedberg E.C., Walker G.C., ea., 1991 ]), а комплементационная
коррекция возможных мутантных линий клеток китайского хомячка, чувствительных к УФ-свету, способствовала выявлению дополнительных генов системы ЭРН. Последние получили название гены ERCC (excision repair cross-complimenting), и поэтому в названии генов и белков ЭРН используются обе аббревиатуры. Хотя детали процесса ЭРН у человека не ясны, так как не определена роль целого ряда генов, в первом приближении он выглядит так ( табл. 3 ) [ Lehmann A.R., 1995 , Lindahl T., ea, 1997 , Hoeijmakers J., 1993 ]. Белок ХРА в комплексе с онДНК-связующим белком RPA ( репликационным белком А ), транслоцируясь вдоль онДНК, опознает конформационнoе повреждениe. Взаимодействуя через другой участок белка ХРА с базальным фактором транскрипции TFIIH (две из субъединиц которого, белки ХРВ и ХРD , обладают геликазной активностью с противоположной ориентацией раскручивания днДНК), они образуют комплекс, который расплетает ДНК вокруг повреждения (в состав этого комплекса входит и белок ХРС
с неясными функциями). Через третий участок белка ХРА к комплексу примыкает гетеродимер ERCC1-XPF , который вносит однонитевой разрыв в ДНК с 5′-конца на расстоянии 16-25 н. от повреждения, тогда как белок XPG , входящий в комплекс белков эксцинуклеазы через взаимодействие с белком RPA, делает надрез с 3′-конца на расстоянии 2-9 н. (в разрезании принимает участие и белок ХРС , а белок ХРЕ активирует реакцию). В результате бимодальной инцизии участок ДНК размером около 29 н. высвобождается, а образующаяся брешь ресинтезируется с помощью ДНК-полимеразы епсилон или ДНК-полимеразы дельта , сопутствующего репликации фактора PCNА , репликационного фактора C-RFС и ДНК-лигазы I .
Представленная картина во многом основана на реконструировании процесса в открытой системе с участием 10 хорошо очищенных белков [ Wood R.D., 1994 ]. Как видно, эукариотическая система ЭРН лишь функционально подобна прокариотической. В ней задействовано значительно больше белков, число которых должно еще возрасти
за счет белков, осуществляющих разборку и сборку хроматина. Раскрытие природы ТЭРН у человека также связывают с пониманием молекулярного дефекта,приводящего к развитию двух мультисистемных генетических заболеваний — синдрома Кокейна (СS) и трихотиодистрофии ( ТТD ). При обоих заболеваниях соматические клетки больных оказались неспособны к ТЭРН. Классические случаи CS оказались связанными с повреждениями в двух генах, CSA и CSB, а редкие смешанные формы CS+XP (CS с дополнительными симптомами ХР), с повреждениями в генах ХРВ, ХРD и ХРG. При ТТD дефекты были обнаружены также в генах XPB и XPD и в новом пока не клонированном гене ТТDА, продукт которого является частью корового домена транскрипционного фактора TFIIH [ Lehmann A.R., 1995 , Lindahl T., ea, 1997 ].
Фактор TFIIH состоит из 6 субъединиц, две из которых — белки XPB и XPD. Связываясь с белками РНК-полимеразного комплекса, кор TFIIH принимает участие в инициировании транскрипции [ Buratowski S., 1994 ], a вкупе с белками репарационного
комплекса кор этого фактора участвует в ЭРН. Если допустить, что белки CSA и CSB способствуют превращению транскрипционного комплекса в репарационнный [ Lindahl T., ea, 1997 , Bregman D.B., 1996 ], то следствием повреждения этих белков может стать дефектность в ТЭРН. Таким образом, либо прямыми воздействиями на коровый домен фактора TFIIH (мутации в генах XPB, XPD и TTDA), либо косвенными (через гены CSA и CSB) можно модифицировать способность этого фактора к диссоциации из РНК- полимеразного комплекса для участия в ТЭРН. Что касается белка XPG, то он, подобно своему гомологу из S. cereivisiae — белку Rad2 [ Bregman D.B., 1996 ], способен взаимодействовать c TFIIH, что допускает возможность его воздействия на TFIIH для участия в ТЭРН. Таковы гипотетические объяснения взаимосвязи молекулярных дефектов при CS и ТTD с нарушениями ТЭРН, которые могут быть полезными для раскрытия механизма ТЭРН у эукариот. Связь же между молекулярными дефектами и клиническими проявлениями
рассматриваемых наследственных заболеваний пока не имеет даже упрощенных толкований [ Kolodner R., 1995 ].
Так случилось, что изучение молекулярных механизмов ЭРН и ТЭРН на клетках человека развивались параллельно, или даже опережая исследования на популярной эукариотической модели дрожжей S. cerevisiae. В табл. 3 представлены известные функциональные аналоги системы ЭРН у дрожжей. Несомненно, однако, что при исследовании детального механизма ЭРН именно эта модельная система будет играть ведущую роль. Например, немаловажной особенностью системы ЭРН у E. coli является ее связь с SOS-функциями клетки. Действительно, центральные гены системы ЭРН, uvrA и uvrB, находятся под контролем SOS-регулона [ Friedberg E.C., Walker G.C., ea., 1991 ]. Системы, подобной SOS, у эукариот пока не обнаружено. Однако целый ряд генов системы ЭРН у дрожжей, таких, как RAD2, RAD7, RAD23, CDC8 и CDC9, индуцируется при повреждении ДНК, а последние два гена дополнительно регулируются клеточным циклом [ Friedberg
E.C., Walker G.C., ea., 1991 ].
Природу этой индукции еще предстоит выяснить.