Спектроскопические
методы анализа основаны на избирательном
поглощении электромагнитного излучения
анализируемым веществом и служат для
исследования строения, идентификации
и количественного определения
светопоглощающих соединений.
В
зависимости от используемой аппаратуры
в фармацевтическом анализе различают
следующие методы анализа, основанные
на поглощении электромагнитного
излучения и испускании света:
—
спектрофотометрия в ультрафиолетовой
(УФ) и видимой областях;
—
спектрофотометрия в инфракрасной (ИК)
области;
—
атомно-эмиссионная и атомно-абсорбционная
спектроскопия (АЭС и ААС);
—
флуориметрия;
—
спектроскопия ядерного магнитного
резонанса (ЯМР).
Ряд
длин волн, для которых проводятся
измерения методами абсорбционной
спектрофотометрии, охватывает спектральную
область от коротких длин волн в УФ-области
до ИК-области. Для удобства отнесений
этот спектральный ряд делится на
следующие диапазоны длин волн: УФ (от
190 до 380 нм), видимый (от 380 до 780 нм), ИК (от
0,78 до 400 мкм).
12.1. Спектрофотометрия в ультрафиолетовой и
ВИДИМОЙ
ОБЛАСТЯХ (ОФС 42-0042-07)
Уменьшение
величины монохроматического излучения,
проходящего через гомогенную поглощающую
среду, количественно описывается законом
Бугера-Ламберта-Бера:
log10(1/T)
= A = эпсилон x c x b,
(1)
где:
T
— пропускание; T — I/I ;
o
I
— интенсивность прошедшего монохроматического
излучения;
I
— интенсивность падающего монохроматического
излучения;
o
эпсилон
— молярный показатель поглощения;
c
— молярная концентрация вещества в
растворе;
b
— длина оптического пути или толщина
слоя, в сантиметрах.
Величина
log10(1/T) носит название оптической
плотности, обозначается буквой A и
является измеряемой величиной. В
отсутствии других физико-химических
факторов измеренная оптическая плотность
(A) пропорциональна концентрации вещества
в растворе (c) и толщине слоя (b).
1%
Величина
A представляет собой удельный показатель
поглощения, т.е.
1
см
оптическую
плотность раствора вещества с
концентрацией 10 г/л (1 г/100 мл)
1%
в
кювете с толщиной слоя 1 см. Величины
A и эпсилон связаны
1
см
соотношением:
1%
10 x эпсилон
A
= ————-, (2)
1
см М.м.
где
М.м. — молекулярная масса исследуемого
вещества.
Измерение
оптической плотности. Если нет других
указаний в частной статье, измерение
оптической плотности проводят при
указанной длине волны с использованием
кювет с толщиной слоя 1 см и при температуре
(20 +/- 1) град. C по сравнению с тем же
растворителем или той же смесью
растворителей, в которой растворено
вещество. При измерении оптической
плотности раствора при данной длине
волны оптическая плотность кюветы с
растворителем, измеренная против воздуха
при той же длине волны, не должна превышать
0,4 и желательно, чтобы она была менее
0,2. Для снижения величины ошибки при
определении оптической плотности
концентрация раствора (а иногда и толщина
слоя) подбираются таким образом, чтобы
оптическая плотность в исследуемой
спектральной области находилась в
пределах от 0,2 до 0,8.
Спектр
поглощения представляют таким образом,
чтобы оптическая плотность или ее
некоторая функция были приведены по
оси ординат, а длина волны или некоторая
функция длины волны — по оси абсцисс.
Если
в частной статье для максимума поглощения
указывается только одна длина волны,
то это означает, что полученное значение
максимума не должно отличаться от
указанного более чем на +/- 2 нм.
Приборы.
Спектрофотометры, предназначенные для
измерений в ультрафиолетовой и видимой
областях спектра, состоят из оптической
системы, выделяющей монохроматическое
излучение в области от 190 до 780 нм и
обеспечивающей его прохождение через
образец, и устройства для измерения
оптической плотности.
Основными
частями этих приборов являются: источник
излучения, диспергирующий прибор (призма
или решетка), щель для выделения полосы
длин волн, кюветы для образцов, детектор
излучаемой энергии, встроенные усилители
и измерительные приборы.
Проверка
шкалы длин волн в УФ и видимой области.
Точность калибровки прибора по шкале
длин волн в спектральном ряду проверяют
по приведенным в табл. 12.1.1 спектральным
линиям водородной (Hбета) или дейтериевой
(Dбета) разрядной лампы, линиям паров
ртути (Hg) кварцево-ртутной дуговой лампы,
а также по максимумам поглощения раствора
гольмия перхлората (Ho) (готовый реактив
для калибровки спектрофотометра
представляет собой 4% раствор гольмия
оксида в 1,4 М растворе хлорной кислоты).
Допустимое отклонение составляет +/- 1
нм для ультрафиолетовой и +/- 3 нм для
видимой области.
Таблица
12.1.1
Спектральные
линии для проверки шкалы длин волн
|
241,15 |
404,66 |
|
253,7 |
435,83 |
|
287,15 |
486,0 |
|
302,25 |
486,1 |
|
313,16 |
536,3 |
|
334,15 |
546,07 |
|
361,5 |
576,96 |
|
365,48 |
579,07 |
Шкала
длин волн может быть калибрована также
при помощи подходящих стеклянных
фильтров, которые имеют фиксированные
полосы поглощения в видимой и УФ-областях,
а также стандартных стекол, содержащих
дидим (смесь празеодима и неодима), и
стекол, содержащих гольмий.
Проверка
шкалы оптической плотности. Для
проверки шкалы оптической
плотности
используют стандартные неорганические
стеклянные фильтры или
раствор
калия дихромата при длинах волн,
указанных в табл. 12.1.2, где для
каждой
длины волны приведено точное
значение удельного показателя
1%
поглощения
A и допустимые пределы.
1
см
Раствор
калия дихромата готовят следующим
образом:
от
57,0 до 63,0 мг (точная навеска) калия
дихромата, предварительно высушенного
до постоянной массы при температуре
130 град. C, растворяют в 0,005 М растворе
серной кислоты и доводят объем раствора
тем же растворителем до 1000 мл.
Таблица
12.1.2
Удельный
показатель поглощения стандартов при
различных
длинах волн
┌─────────────────────┬─────────────────────────┬─────────────────────────┐
│ Длина
волны, в │ Удельный показатель │
Допустимые пределы │
│ нанометрах
│ поглощения │ 1%
│
│ │ 1%
│ для A │
│ │ A
│ 1 см │
│ │ 1
см │ │
├─────────────────────┼─────────────────────────┼─────────────────────────┤
│ 235
│ 124,5 │ От 122,9 до 126,2 │
├─────────────────────┼─────────────────────────┼─────────────────────────┤
│ 257
│ 144,5 │ От 142,8 до 146,2 │
├─────────────────────┼─────────────────────────┼─────────────────────────┤
│ 313
│ 48,6 │ От 47,0 до 50,3 │
├─────────────────────┼─────────────────────────┼─────────────────────────┤
│ 350
│ 107,3 │ От 105,6 до 109,0 │
└─────────────────────┴─────────────────────────┴─────────────────────────┘
Предельный
уровень рассеянного света. Рассеянный
свет может быть обнаружен при данной
длине волны с использованием соответствующих
фильтров или растворов: например,
оптическая плотность раствора 12 г/л
калия хлорида в кювете с толщиной слоя
1 см при 200 нм при использовании воды в
качестве раствора сравнения должна
быть больше 2.
Разрешающая
способность (для качественного анализа).
Если есть указание в частной статье,
определяют разрешающую способность
спектрофотометра следующим образом.
Записывают спектр 0,02% (об/об) раствора
толуола в гексане. Минимально допустимое
значение отношения оптической плотности
в максимуме поглощения при 269 нм к
оптической плотности в минимуме
поглощения при 266 нм указывают в частной
статье.
Ширина
спектральной щели (для количественного
анализа). В случае использования
спектрофотометра с изменяемой шириной
спектральной щели при выбранной длине
волны возможны погрешности, связанные
с шириной этой щели. Для их исключения
ширина щели должна быть малой по сравнению
с полушириной полосы поглощения (шириной
на половине оптической плотности) и в
то же время должна быть максимально
велика для получения высокого значения
интенсивности падающего монохроматического
излучения (Io). Таким образом, ширина щели
должна быть такой, чтобы дальнейшее ее
уменьшение не изменяло величину
измеряемой оптической плотности.
Кюветы.
Допустимые отклонения в толщине слоя
используемых кювет должны быть не более
+/- 0,005 см. Кюветы, предназначенные для
испытуемого раствора и раствора
сравнения, должны иметь одинаковое
пропускание (или оптическую плотность)
при заполнении одним и тем же растворителем.
В противном случае это различие следует
учитывать.
Требования
к растворителям. Для определений,
производимых в ультрафиолетовой и
видимой областях, образец анализируемого
вещества растворяют в соответствующем
растворителе, который должен быть
оптически прозрачным в используемой
области длин волн. Для этих областей
длин волн пригодны многие растворители,
в том числе вода, спирты, хлороформ,
низшие углеводороды, эфиры и разбавленные
растворы сильных кислот и щелочей.
Идентификация
Абсорбционную
спектрофотометрию в ультрафиолетовой
и видимой областях спектра применяют
для определения подлинности лекарственных
средств путем:
—
сравнения спектров поглощения испытуемого
раствора и раствора стандартного
образца; в указанной области спектра
должно наблюдаться совпадение положений
максимумов, минимумов, плеч и точек
перегиба;
—
указания положений максимумов, минимумов,
плеч и точек перегиба; расхождение между
наблюдаемыми и указанными длинами волн
в максимумах и минимумах поглощения не
должно обычно превышать +/- 2 нм.
Возможны
и другие варианты применения, оговоренные
в частных фармакопейных статьях.
Количественное
определение
Определение
концентрации веществ спектрофотометрическим
методом основано на использовании
закона Бугера-Ламберта-Бера в форме:
A
C
= ———-, (3)
1%
A
x b
1
см
где:
C
— концентрация вещества в г/100 мл;
A
— оптическая плотность испытуемого
раствора;
1%
A
— удельный показатель поглощения
вещества;
1
см
b
— толщина поглощающего слоя, в сантиметрах.
В
ряде случаев даже при использовании
монохроматического излучения могут
наблюдаться отклонения от закона
Бугера-Ламберта-Бера, обусловленные
процессами диссоциации, ассоциации и
комплексообразования. Поэтому
предварительно следует проверить
линейность зависимости оптической
плотности раствора от концентрации в
аналитической области. При наличии
отклонений от линейной зависимости
следует пользоваться не формулой (3), а
экспериментально найденной зависимостью.
Обычно
определение концентрации
спектрофотометрическим методом проводят
с использованием стандартного образца.
Расчет концентрации основан на
использовании уравнения:
C
A
—
= —, (4)
C
A
o
o
где:
C
и C — концентрации испытуемого раствора
и раствора стандартного
o
образца соответственно;
A
и A — оптические плотности испытуемого
раствора и раствора
o
стандартного образца соответственно.
Вначале
измеряют оптическую плотность раствора
стандартного образца, приготовленного,
как указано в частной фармакопейной
статье, затем проводят измерение
оптической плотности испытуемого
раствора. Второе измерение проводят
сразу после первого, с использованием
той же кюветы, в тех же экспериментальных
условиях.
Метод
с использованием стандартного образца
является более точным и надежным.
Возможность применения значения
удельного показателя поглощения в
каждом конкретном случае следует
обосновывать. Обычно метод с использованием
значения удельного показателя поглощения
применим при допусках содержания
анализируемого вещества не менее +/- 10%
от номинального содержания.
Многокомпонентный
спектрофотометрический анализ
Многокомпонентный
спектрофотометрический анализ (анализ
смесей) применяют для одновременного
количественного определения нескольких
компонентов лекарственных средств,
каждое из которых подчиняется закону
Бугера-Ламберта-Бера.
Количественное
определение в многокомпонентном
спектрофотометрическом анализе
основывается обычно на использовании
уравнения:
m
А
= SUM E x c i = 1,…n, (5)
i
j = 1 ij j
где:
А
— оптическая плотность испытуемого
раствора при i-ой длине волны;
i
E
— показатели поглощения (зависящие
от способа выражения
Ij
концентрации)
j-го компонента образца при i-ой
аналитической длине волны;
c
— концентрация j-го компонента образца.
j
Соответствующие
методики проведения анализа и расчетные
формулы указываются в частных фармакопейных
статьях.
Производная
спектрофотометрия
В
производной спектрофотометрии исходные
спектры поглощения (нулевого порядка)
преобразуются в спектры производных
первого, второго и более высокого
порядков.
Спектр
первой производной представляет собой
график зависимости градиента кривой
поглощения (скорость изменения оптической
плотности с длиной волны, d A/d лямбда) от
длины волны.
Спектр
второй производной представляет
собой график зависимости
2
2
кривизны
спектра поглощения (d А/d лямбда ) от
длины волны. Вторая
производная
при любой длине волны связана с
концентрацией следующим
соотношением:
2
1%
2
d A
d
A 1 см
———
= ———- x c x l, (6)
2
2
d
лямбда d лямбда
где:
A
— оптическая плотность при длине волны
лямбда;
1%
A
— удельный показатель поглощения при
длине волны лямбда;
1
см
c
— концентрация вещества в растворе, в
граммах/100 мл;
l
— толщина слоя, в сантиметрах.
Производная
спектрофотометрия может быть использована
как для целей идентификации веществ,
так и их количественного определения
в многокомпонентных смесях, а также в
тех случаях, когда имеется фоновое
поглощение, вызванное присутствием
веществ, содержание которых не
регламентируется.
Приборы.
Используют спектрофотометры, отвечающие
указанным выше требованиям и оснащенные
аналоговым резистентно-емкостным
дифференцирующим модулем или цифровым
дифференциатором, или другими средствами
получения производных спектров, в
соответствии с инструкцией к прибору.
Некоторые методы получения спектров
второй производной приводят к смещению
длин волн относительно исходного
спектра, что следует учитывать там, где
это необходимо.
Разрешающая
способность. Если указано в частных
фармакопейных статьях, записывают
спектр второй производной для раствора
0,2 г/л толуола в метаноле, используя
метанол в качестве раствора сравнения.
На спектре должен присутствовать
небольшой отрицательный экстремум,
расположенный между двумя большими
отрицательными экстремумами при 261 нм
и 268 нм, в соответствии с рис. 12.1.1. (не
приводится). Если нет других указаний
в частных фармакопейных статьях,
отношение A/B должно быть не менее 0,2.
Методика.
Процедура анализа аналогична применяемой
в обычной спектрофотометрии, но вместо
оптических плотностей используют
производные. Готовят раствор испытуемого
образца, настраивают прибор в соответствии
с инструкцией производителя и рассчитывают
количество определяемого вещества, как
указано в частной фармакопейной статье.
Рис.
12.1.1. Спектр второй производной раствора
толуола
(0,2 г/л) в метаноле
Рисунок
не приводится.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Оптическая плотность раствора
Колориметрия
Из оптических методов анализа в практике аналитических лабораторий наиболее широко применяются колориметрические методы (от лат. color — цвет и греч. μετρεω — измеряю). Колориметрические методы основаны на измерении интенсивности светового потока, прошедшего через окрашенный раствор.
В колориметрическом методе используются химические реакции, сопровождающиеся изменением цвета анализируемого раствора. Измеряя светопоглощение такого окрашенного раствора или сравнивая полученную окраску с окраской раствора известной концентрации, определяют содержание окрашенного вещества в испытуемом растворе.
Существует зависимость между интенсивностью окраски раствора и содержанием в этом растворе окрашенного вещества. Эта зависимость, называемая основным законом светопоглощения (или законом Бугера—Ламберта—Бера), выражается уравнением:
I = I0 10 — ε c l
где I — интенсивность света, прошедшего через раствор; I0 — интенсивность падающего на раствор света; ε- коэффициент светопоглощения, постоянная величина для каждого окрашенного вещества, зависящая от его природы; С — молярная концентрация окрашенного вещества в растворе; l — толщина слоя светопоглощающего раствора, см.
Физический смысл этого закона можно выразить следующим образом. Растворы одного и того же окрашенного вещества при одинаковой концентрации этого вещества и толщине слоя раствора поглощают равное количество световой энергии, т. е. светопоглощение таких растворов одинаковое.
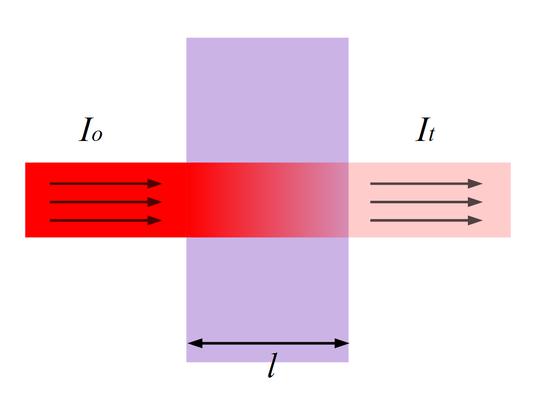
Для окрашенного раствора, заключенного в стеклянную кювету с параллельными стенками, можно сказать, что по мере увеличения концентрации и толщины слоя раствора его окраска увеличивается, а интенсивность света I, прошедшего через поглощающий раствор, уменьшается по сравнению с интенсивностью падающего света I0.
Рис.1 Прохождение света через кювету с исследуемым раствором.
Оптическая плотность раствора.
Если прологарифмировать уравнение основного закона светопоглощения и изменить знаки на обратные, то уравнение принимает вид:

Величина  является очень важной характеристикой окрашенного раствора; ее называют оптической плотностью раствора и обозначают буквой A:
является очень важной характеристикой окрашенного раствора; ее называют оптической плотностью раствора и обозначают буквой A:
A = ε C l
Из этого уравнения вытекает, что оптическая плотность раствора прямо пропорциональна концентрации окрашенного вещества и толщине слоя раствора.
Другими словами, при одинаковой толщине слоя раствора данного вещества оптическая плотность этого раствора будет тем больше, чем больше в нем содержится окрашенного вещества. Или, наоборот, при одной и той же концентрации данного окрашенного вещества оптическая плотность раствора зависит только от толщины его слоя. Отсюда может быть сделан следующий вывод: если два раствора одного и того же окрашенного вещества имеют различную концентрацию, одинаковая интенсивность окраски этих растворов будет достигнута при толщинах их слоев, обратно пропорциональных концентрациям растворов. Этот вывод очень важен, так как на нем основаны некоторые методы колориметрического анализа.
Таким образом, чтобы определить концентрацию (С) окрашенного раствора, необходимо измерить его оптическую плотность (A). Чтобы измерить оптическую плотность, следует измерить интенсивность светового потока.
Интенсивность окраски растворов можно измерять различными методами. Различают субъективные (или визуальные) методы колориметрии и объективные (или фотоколориметрические).
Визуальными называются такие методы, при которых оценку интенсивности окраски испытуемого раствора делают невооруженным глазом.
При объективных методах колориметрического определения для измерения интенсивности окраски испытуемого раствора вместо непосредственного наблюдения пользуются фотоэлементами. Определение в этом случае проводят в специальных приборах — фотоколориметрах, откуда и метод получил название фотоколориметрического.
Визуальные методы
К визуальным методам относятся:
1) метод стандартных серий;
2) метод дублирования (колориметрическое титрование);
3) метод уравнивания.
Метод стандартных серий. При выполнении анализа методом стандартных серий интенсивность окраски анализируемого окрашенного раствора сравнивают с окрасками серии специально приготовленных стандартных растворов (при одинаковой толщине поглощающего слоя).
Растворы в колориметрии обычно имеют интенсивную окраску, поэтому имеется возможность определять весьма небольшие концентрации или количества веществ. Однако это может сопровождаться определенными трудностями: так навески для приготовления серии стандартных растворов могут быть очень малы. Для преодоления этих трудностей готовят стандартный раствор А достаточно высокой концентрации, например 1 мг/мл. После этого путем разбавления из раствора А готовят стандартный раствор В значительно меньшей концентрации, а из него в свою очередь готовят серию стандартных растворов.
Для этого в пробирки или кюветы одинакового размера и одинакового цвета стекла пипеткой добавляются необходимые объемы растворов реагентов в нужной последовательности. Порции растворов определяемого вещества целесообразно добавлять из бюретки, т.к. их объемы будут различны для обеспечения различных концентраций в серии стандартных растворов. При этом начальный раствор должен содержать все компоненты, кроме определяемого вещества (нулевой раствор). В исследуемый раствор добавляют растворы необходимых реагентов. Все растворы доводят до постоянного объема, а затем визуально сравнивают интенсивность окраски исследуемого раствора с растворами серии стандартных растворов. Возможно совпадение интенсивности окраски с каким-либо раствором серии. Тогда считается, сто исследуемый раствор имеет такую же концентрацию или содержит столько же определяемого вещества. Если же интенсивность окраски покажется промежуточной между соседними растворами серии, концентрация или содержание определяемого компонента считают средним арифметическим между растворами серии.
Колориметрическое титрование (метод дублирования). Этот метод основан на сравнении окраски анализируемого раствора с окраской другого раствора— контрольного. Для приготовления контрольного раствора готовят раствор, содержащий все компоненты исследуемого раствора, за исключением определяемого вещества, и все употреблявшиеся при подготовке пробы реактивы, и к нему добавляют из бюретки стандартный раствор определяемого вещества. Когда этого раствора будет добавлено столько, что интенсивности окраски контрольного и анализируемого раствора уравняются, считают, что в анализируемом растворе содержится столько же определяемого вещества, сколько его было введено в контрольный раствор.
Метод уравнивания.Этот метод основан на уравнивании окрасок анализируемого раствора и раствора с известной концентрацией определяемого вещества — стандартного раствора. Существуют два варианта выполнения колориметрического определения этим методом.
По первому варианту уравнивание окрасок двух растворов с разной концентрацией окрашенного вещества проводят путем изменения толщины слоев этих растворов при одинаковой силе проходящего через растворы светового потока. При этом, несмотря на различие концентраций анализируемого и стандартного растворов, интенсивность светового потока, проходящего через оба слоя этих растворов, будет одинакова. Соотношение между толщинами слоев и концентрациями окрашенного вещества в растворах в момент уравнивания окрасок будет выражаться уравнением:
где l1 — толщина слоя раствора с концентрацией окрашенного вещества C1, а l2-толщина слоя раствора с концентрацией окрашенного вещества C2.
В момент равенства окрасок отношение толщин слоев двух сравниваемых растворов обратно пропорционально отношению их концентраций.
На основании приведенного уравнения, измерив толщину слоев двух одинаково окрашенных растворов и зная концентрацию одного из этих растворов, легко можно рассчитать неизвестную концентрацию окрашенного вещества в другом растворе.
Для измерения толщины слоя, через который проходит световой поток, можно применять стеклянные цилиндры или пробирки, а при более точных определениях специальные приборы — колориметры.
По второму варианту, для уравнивания окрасок двух растворов с различной концентрацией окрашенного вещества, через слои растворов одинаковой толщины пропускают световые потоки различной интенсивности.
В этом случае оба раствора имеют одинаковую окраску, когда отношение логарифмов интенсивностей падающих световых потоков равно отношению концентраций.
В момент достижения одинаковой окраски двух сравниваемых растворов, при равной толщине их слоев, концентрации растворов прямо пропорциональны логарифмам интенсивностей падающего на них света.
По второму варианту определение может быть выполнено только с помощью колориметра.
Теоретические основы определения оптической плотности раствора

Любая частица, будь то молекула, атом или ион, в результате поглощения кванта света переходит на более высокий уровень энергетического состояния. Чаще всего осуществляется переход из основного в возбужденное состояние. Это вызывает появление в спектрах определенных полос поглощения.
Поглощение излучения приводит к тому, что при пропускании его через вещество интенсивность этого излучения снижается при увеличении количества частиц вещества, обладающего некоторой оптической плотностью. Этот метод исследования предложил В. М. Севергин еще в 1795 году.
Наилучшим образом этот метод годится для реакций, где определяемое вещество способно переходить в окрашенное соединение, что вызывает изменение окраски исследуемого раствора. Измерив его светопоглощение или сравнив окраску с раствором известной концентрации, несложно найти процент содержания вещества в растворе.

Основной закон светопоглощения
Суть фотометрического определения заключается в двух процессах:
- перевод определяемого вещества в поглощающее электромагнитные колебания соединение;
- замер интенсивности поглощения этих самых колебаний раствором исследуемого вещества.
Изменения в интенсивности потока света, проходящего через светопоглощающее вещество, будут вызываться также потерями света из-за отражения и рассеяния. Чтобы результат был достоверным, проводят параллельные исследования по замеру параметров при той же толщине слоя, в идентичных кюветах, с тем же растворителем. Так снижение интенсивности света зависит главным образом от концентрации раствора.
Уменьшение интенсивности света, пропущенного через раствор, характеризуют коэффициентом светопропускания (также принято называть его пропусканием) Т:
- I — интенсивность света, пропущенного через вещество;
- I0 — интенсивность падающего пучка света.
Таким образом, пропускание показывает долю непоглощенного светового потока, проходящего через изучаемый раствор. Обратный алгоритм значения пропускания называют оптической плотностью раствора (D): D = (-lgT) = (-lg) * (I / I0) = lg * (I0 / I).
Это уравнение показывает, какие параметры являются главными для исследования. К ним относится длина волны света, толщина кюветы, концентрация раствора и оптическая плотность.
Закон Бугера-Ламберта-Бера
Он является математическим выражением, отображающим зависимость уменьшения интенсивности монохроматического потока света от концентрации светопоглощающего вещества и толщины жидкостного слоя, через который он пропущен:
I = I0 * 10 -ε·С·ι , где:
- ε — коэффициент поглощения света;
- С — концентрация вещества, моль/л;
- ι —толщина слоя анализируемого раствора, см.
Преобразовав, эту формулу можно записать: I / I0 = 10 -ε·С·ι .
Суть закона сводится к следующему: различные растворы одного и того же соединения при равной концентрации и толщине слоя в кювете поглощают одинаковую часть падающего на них света.
Прологарифмировав последнее уравнение, можно получить формулу: D = ε * С * ι.
Очевидно, что оптическая плотность напрямую зависит от концентрированности раствора и толщины его слоя. Становится ясен физический смысл молярного коэффициента поглощения. Он равен D для одномолярного раствора и при толщине слоя в 1 см.

Ограничения применения закона
Этот раздел включает следующие пункты:
- Он справедлив исключительно для монохроматического света.
- Коэффициент ε связан с показателем преломления среды, особенно сильные отклонения от закона могут наблюдаться при анализе высококонцентрированных растворов.
- Температура при измерении оптической плотности должна быть постоянной (в рамках нескольких градусов).
- Световой пучок должен быть параллельным.
- рН среды должен быть постоянным.
- Закон применим для веществ, светопоглощающими центрами которых являются частицы одного вида.
Методы определения концентрации
Стоит рассмотреть метод градуировочного графика. Для его построения готовят ряд растворов (5-10) с различной концентрацией исследуемого вещества и замеряют их оптическую плотность. По полученным значениям выстраивают график зависимости D от концентрации. График является прямой линией, идущей от начала координат. Он позволяет легко определить концентрацию вещества по результатам проведенных измерений.
Также существует метод добавок. Применяется реже, чем предыдущий, но позволяет проанализировать растворы сложного состава, поскольку учитывает влияние дополнительных компонентов. Суть его состоит в определении оптической плотности среды Dx, содержащей определяемое вещество неизвестной концентрации Сх, с повторным анализом того же раствора, но с добавлением определенного количества исследуемого компонента (Сст). Величину Сх находят, используя расчеты или графики.

Условия проведения исследования
Чтобы фотометрические исследования давали достоверный результат, необходимо соблюдать несколько условий:
- реакция должна заканчиваться быстро и полностью, избирательно и воспроизводимо;
- окраска образующегося вещества должна быть устойчива во времени и не изменяться под действием света;
- исследуемое вещество берут в количестве, которого достаточно для перевода его в аналитическую форму;
- замеры оптической плотности проводят в том интервале длин волн, при котором различие в поглощении исходных реагентов и анализируемого раствора наибольшее;
- светопоглощение раствора сравнения принято считать оптическим нулем.
Способы расчета концентрации по величине аналитического сигнала

СПОСОБЫ РАСЧЕТА КОНЦЕНТРАЦИИ ПО ВЕЛИЧИНЕ АНАЛИТИЧЕСКОГО СИГНАЛА
МЕТОД ГРАДУИРОВОЧНОГО ГРАФИКА
Пример 1. При измерении оптической плотности в одинаковых условиях (длина волны 340 нм, толщина поглощающего слоя – 1,00 см) растворов калия дихромата с разной концентрацией хрома (мкг/мл) получены следующие результаты:
Изобразите примерный вид градуировочного графика; методом наименьших квадратов рассчитайте обратное уравнение градуировочного графика с = bA + а; определите концентрацию хрома (мкг/мл) в растворе Х, имеющем оптическую плотность 0,480.
Рассчитайте массу хрома (мг) в анализируемой пробе, если ее растворили в присутствии концентрированной серной кислоты в воде дистиллированной в мерной колбе объёмом 50,00 мл (раствор Х).
1. Расчет методом наименьших квадратов

2. Расчет с помощью программы Excel

длина волны 340 нм
Ответ: с = 115,72А — 8,8397 (r = 0,9941); масса хрома в пробе 2,34 мг
Пример 2. При измерении оптической плотности в одинаковых условиях (длина волны 400 нм, толщина поглощающего слоя – 1,00 см) растворов никеля (II) нитрата с разной концентрацией никеля (мг/мл) получены следующие результаты:
Изобразите примерный вид градуировочного графика; методом наименьших квадратов рассчитайте обратное уравнение градуировочного графика с = bA + а; определите концентрацию никеля (мг/мл) в растворе Х, имеющем оптическую плотность 0,350.
Рассчитайте массу никеля (мг) в анализируемой пробе, если ее количественно перенесли в мерную колбу объёмом 25,00 мл и развели водой дистиллированной в присутствии азотной кислоты до метки (раствор Х).
1. Расчет методом наименьших квадратов

2. Расчет с помощью программы Excel

Ответ: с = 42,495А — 3,8535 (r = 0,9986); масса никеля 275 мг
Пример 3. При измерении оптической плотности в одинаковых условиях (длина волны 620 нм, толщина поглощающего слоя – 1,00 см) растворов меди (II) в виде аммиачного комплекса с разной концентрацией меди (мкг/мл) получены следующие результаты:
Изобразите примерный вид градуировочного графика; методом наименьших квадратов рассчитайте обратное уравнение градуировочного графика с = bA; определите концентрацию меди (мкг/мл) в растворе Х, имеющем оптическую плотность 0,150.
Рассчитайте массу меди (мкг) в анализируемой пробе, если ее количественно перенесли в мерную колбу объёмом 25,00 мл и до метки развели водой дистиллированной в присутствии избытка аммиака (раствор Х).
1. Расчет методом наименьших квадратов
2. Расчет с помощью программы Excel

2. Расчет с помощью программы Excel

Ответ: с = 34,595А (r = 0,9985); масса меди 130 мкг.
МЕТОД ОДНОГО СТАНДАРТНОГО РАСТВОРА
Измеряют величину аналитического сигнала (yст) для раствора с известной концентрацией вещества (сст). Затем измеряют величину аналитического сигнала (yx) для раствора с неизвестной концентрацией вещества (сx). Такой способ расчёта можно использовать в том случае, если зависимость аналитического сигнала от концентрации описывается линейным уравнением без свободного члена. Концентрация вещества в стандартном растворе должна быть такой, чтобы величины аналитических сигналов, полученных при использовании стандартного раствора и раствора с неизвестной концентрацией вещества, были бы как можно ближе друг к другу.

ПРИМЕР 1. При фотометрическом определении концентрации нитрит-ионов с помощью реактива Грисса (раствора сульфаниловой кислоты и α-нафтиламина в разбавленной уксусной кислоте) было установлено, что раствор с концентрацией нитрит-ионов 2,00 мкг/мл имеет в соответствующих условиях оптическую плотность 0,300. Рассчитайте концентрацию нитрит-ионов в растворе (мкг/мл), оптическая плотность которого в таких же условиях равна 0,250. Зависимость оптической плотности от содержания аналита линейна и проходит через начало координат.

Ответ: 1,67 мкг/мл
МЕТОД ДВУХ СТАНДАРТНЫХ РАСТВОРОВ
(метод ограничивающих растворов)
Измеряют величины аналитических сигналов для стандартных растворов с двумя разными концентрациями вещества, одна из которых (с1) меньше предполагаемой неизвестной концентрации (сx), а вторая (с2) – больше. Его используют, если зависимость аналитического сигнала от концентрации описывается линейным уравнением, не проходящим через начало координат.
Пример 1. Раствор с концентрацией никеля (II) 12,00 мг/мл имеет оптическую плотность 0,350 нм, а с концентрацией 16,00 мг/мл – 0,440. Определите концентрацию никеля (мг/мл) в растворе с оптической плотностью 0,380 (все измерения проводились в одинаковых условиях: длина волны 400 нм, толщина поглощающего слоя – 1,00 см, раствор в азотной кислоте).

Ответ: 13,33 мг/мл
Используют при анализе сложных матриц, когда матричные компоненты оказывают влияний на величину аналитического сигнала и невозможно точно скопировать матричный состав образца, в случае линейной зависимости, проходящей через начало координат.
Вначале измеряют величину аналитического сигнала (yx) для пробы с неизвестной концентрацией вещества. Затем к данной пробе прибавляют некоторое точное количество определяемого вещества (стандарта) и снова измеряют величину аналитического сигнала (yдоб). Концентрацию определяемого компонента в анализируемой пробе (без учета разбавления) рассчитывают по формуле:

Для учета разбавления раствора используем формулу:

ПРИМЕР 1. Раствор с неизвестной концентрацией вещества имел оптическую плотность 0,300. К 5,00 мл такого раствора прибавили 2,00 мл раствора с концентрацией этого же вещества 40,0 мг/л. Оптическая плотность полученного раствора при измерении её в таких же условиях оказалась равна 0,500. Рассчитайте концентрацию вещества (мг/л) в исходном растворе.
1 способ: пропорционально

2 способ: преобразуем составленную пропорцию в приведенную ранее формулу


ПРИМЕР 2. Оптическая плотность раствора с неизвестным содержанием вещества равна 0,400. При добавлении к анализируемому раствору 10,0 мкг этого же вещества оптическая плотность увеличилась до 0,500. Рассчитайте массу определяемого вещества (мкг) в исходном растворе.
1 способ: пропорционально

2 способ: преобразуем составленную пропорцию в приведенную ранее формулу
источники:
http://fb.ru/article/378024/teoreticheskie-osnovyi-opredeleniya-opticheskoy-plotnosti-rastvora
http://pandia.ru/text/80/260/2737.php
Ошибки спектрофотометрических измерений определяются флуктуациями показаний на выходном приборе. Их величина зависит, в свою очередь, от стабильности источника света, флуктуаций светового пучка на пути от источника света к приемнику, шумов приемно-усилительной аппаратуры и регистрирующего прибора. Рассмотрим влияние этих источников ошибок на результаты измерений, учитывая, что при абсорбционных измерениях, в конечном итоге, существенна точность определения оптической плотности О, а не интенсивностей поглощенного и непоглощенного сигналов. Напомним, что [c.137]
Пусть чувствительность определения примесей в пробе объемом 25 мл при помощи кюветы толщиной 5 см равна 0,005 у/см . Тогда минимальное обнаруживаемое количество вещества равно 0,005 X (25/5) == 0,025 мкг, или 2,5 10 %, для пробы весом 1 г. Эта величина примерно соответствует ошибке спектрофотометрических измерений малых поглощений. [c.133]
Рассмотренные ошибки спектрофотометрического метода в основном относятся к работе прибора. Естественно, что не меньшее значение могут иметь ошибки, связанные с работой самого исследователя (точность приготовления исходных растворов, способ заполнения кювет), и с условиями протекания конкретной химиче- ской реакции (разложение реагентов, межмолекулярные взаимодействия и т. п.). Все это должно учитываться при проведении фотометрических измерений. [c.22]
Изучение факторов, влияющих на точность спектрофотометрических измерений [19] — [27], показывает, что причины ошибок в спектрофотометрии могут быть весьма разнообразны и многочисленны. Ошибки возникают, например, за счет действий оператора, условий проведения реакций, недостаточной чистоты кювет, непостоянства их установки в кюветные отделения, невоспроизводимости настройки шкалы прибора на О и 100% пропускания, непостоянства излучения источника освещения, нестабильности работы фотоэлектрической системы [24] — [27]. [c.30]
При определении Do (ВаО) на основании данных по реакций образования ВаО получаемые значения существенно зависят от принятого в расчете типа основного состояния этой молекулы. Значение, приведенное по данным работ [72, 74, 75, 396], получено для основного состояния Х 2 оно подтверждено результатами измерений методом электронного удара [73]. Расчет по результатам измерения давления пара ВаО менее надежен из-за неточности данных по АЯ (ВаО, тв.) и АЯд (Ва, тв.). В обзоре [76] принимается основное состояние и рекомендуется Do = 131 6. См. также [4, стр. 236]. Спектрофотометрические измерения [118, 119], приведшие к значению —120, содержали ошибки, см. [396]. [c.49]
На абсолютную и относительную точность (воспроизводимость) спектрофотометрических измерений влияет ряд разнообразных и часто трудно поддающихся учету факторов [23, 40]. Для количественного анализа и различных сравнительных исследований наиболее важной является воспроизводимость измерений и несущественны некоторые ошибки систематического характера, так что при разработке многих методик исследования, а также аппаратуры, исключению последних уделяется мало внимания. В связи с этим существует такое положение, что при высокой в большинстве случаев относительной точности современных спектрофотометрических измерений данные, полученные на различных приборах или в различных условиях эксперимента, часто значительно различаются. В большей части опубликованных исследований ультрафиолетовых спектров поглощения авторами не оценивается абсолютная точность измерений, а также не приводятся данные, относящиеся к аппаратуре и методике эксперимента, позволяющие провести хотя бы грубую оценку подобного рода. [c.383]
Расчет общей (максимальной) ошибки и отдельных составляющих ошибок дифференциальных спектрофотометрических измерений кобальта (14—26 г/л) в виде перхлората, измеренных по отношению к оптимальному раствору сравнения, содержащему 12 г/л Со +, приведены в табл. 7, [c.54]
Из уравнения (28) можно также сделать следующие общие заключения по точности дифференциальных спектрофотометрических измерений. Ошибка будет меньше, если [c.44]
Погрешность в определении истинной константы диссоциации слагается в основном из трех величин погрешности, вносимой спектрофотометрическими измерениями, погрешности потенциометрических измерений (если они производились) и ошибки, которая вносится принятой методикой нахождения истинной константы диссоциации по найденному значению кажущейся или концентрационной константы диссоциации. [c.93]
Ошибки при спектрофотометрических измерениях [c.137]
Первоначально рассмотрим точность анализов. Ошибка воспроизводимости, которой характеризуется точность определений атомно-абсорбционного анализа, складывается за счет двух основных операций получения поглощающего слоя и измерения поглощения спектрофотометром. Источники возникновения и величина ошибок при спектрофотометрических измерениях обсуждались ранее ( 19), причем было показано, что эти ошибки могут быть в принципе уменьшены до дробовых шумов приемника, имеющих статистическое происхождение. Случайные ошибки, связанные с получением поглощающего слоя, обусловлены следующими звеньями анализа неоднородностью образцов, дозированием проб на электрод, случайными обстоятельствами, определяющими скорость испарения элемента в кювету, колебаниями температуры кюветы и давления постороннего газа. [c.329]
Непосредственная погрешность спектрофотометрического измерения, включающая ошибки настройки прибора на О и 100% пропускания [76, 83, 88—91], погрешность отсчета по измерительному потенциометру и ошибки, связанные с нестабильностью электронной схемы [82, 92] в процессе измерения. [c.17]
Выбор условий регистрации спектров оказывает существенное влияние на результаты спектрофотометрических измерений. При выборе оптимальных условий удается свести к минимуму как систематические, так и случайные ошибки, возникающие [c.19]
Зависимость ошибки определения концентрации от величины пропускаемости света измеряемым раствором. Одна из существенных ошибок спектрофотометрических измерений возникает при отсчетах величины пропускания на крайних участках соответствующей шкалы прибора. Вследствие логарифмической формы закона поглощения наибольшая точность может быть получена при измерениях в области средних значений величины пропускания. Если предельное значение неопределенности при отсчете О величины отклонения гальванометра обозначить через (10 и предположить, что источник излучения является стабильным и что соотношение между интенсивностью падающего светового пучка и отклонением гальванометра является линейным, то [c.85]
Одновременное присутствие в растворе последовательно образующихся комплексов может привести к серьезным ошибкам при сочетании спектрофотометрических измерений с экстракцией растворителем. Только один из последовательных комплексов — электронейтральный — может экстрагироваться органическим растворителем. Различные последовательно образующиеся комплексы, которые содержат большее или меньшее количество лигандов, чем необходимо для нейтрализации положительного заряда центрального иона металла, и сами несут положительный или отрицательный заряд, останутся в водной фазе. Возможность ошибки будет наименьшей в случае комплексов, в которых для нейтрализации электрического заряда центрального иона металла и насыщения его координационной сферы требуется такое же число лигандов, как и для координации. [c.88]
Дифференциальный спектрофотометрический метод может быть применен и в тех случаях, когда имеется наложение в спектрах поглощения соединения и реагента. Тогда при измерении по отношению к одному из использованных как эталон растворов в значительной степени исключается ошибка за счет поглощения реагента. [c.480]
XI1-3-1. При каком значении Т (парциальное поглощение) в спектрофотометрических анализах будет минимальной относительная ошибка в определении концентрации (Дс/с) для данной ошибки измерения Г [c.143]
Дифференциальный метод анализа используют для повышения точности спектрофотометрических и фотоколориметрических измерений при определении высоких концентраций веществ (от 10 до 100%). Сущность метода заключается в измерении светопоглощения анализируемого раствора относительно раствора сравнения, содержащего определенное количество испытуемого вещества это приводит к изменению рабочей области шкалы прибора и снижению относительной ошибки анализа до 0,5—1%. [c.40]
Кроме того, в настоящее время разработаны спектрофотометрические методы определения большого содержания отдельных компонентов. Эти методы называют дифференциальной фотометрией. Для точного измерения в параллельном световом потоке устанавливают стандартный раствор, близкий по составу к испытуемому раствору. Таким образом, измеряется разница интенсивности двух световых потоков ошибка измерений меньше сказывается на конечном результате. Главные трудности и недостатки, по сравнению с эмиссионным спектральным анализом, связаны с затратой времени на подготовку вещества к анализу, отделение мешающих компонентов, и др. Результат зависит от выбора условий, реактивов и концентрации посторонних ионов. Групповые методы почти не разработаны, поэтому для каждого элемента необходим отдельный ход анализа. [c.9]
Другим методом определения констант является измерение возрастания растворимости в воде исследуемого вещества при различных значениях pH раствора (глава 6), Этот метод не так точен, как потенциометрический, спектрофотометрический и кондуктометрический, но бывает полезен в тех случаях (к счастью, редких), когда вещество слишком мало растворимо в воде для того, чтобы использовать потенциометрический или кондуктометрический метод, и спектр его непригоден для определения. Катализ гидролиза эфиров, дисахаридов и глюкозидов как метод измерения констант ионизации представляет лишь исторический интерес. В ряде случаев этот метод приводил к очень грубым ошибкам. [c.18]
Обычно спектрофотометрические измерения проводят в таких условиях, когда оптическая плотность исследуемого раствора лежит в 1феде-лах А = 0,2—0,8, так как именно при таких значениях оптической плотности достигается минимальная ошибка спектрофотометрических измерений. [c.528]
Общепринято, что такое ограничение не распространяется па спектрофотометрические данные, и отклонения обычно определяют как разность между необходимым и рассчитанным све-топоглощением [4, И, 12, 53, 72—74]. Обычно при определенных условиях нет необходимости использовать веса, так как в этом случае ошибки спектрофотометрического измерения преобладают над ошибками измерения концентрации [11, 12, 75]. Кроме того, показания современных спектрофотометров имеют постоянную дисперсию в некотором диапазоне значений светопоглощения (см. разд. 8.3, п. 6). Однако если измерять светопоглощение одного и того же раствора при нескольких длинах волн, то будет наблюдаться корреляция ошибок. Для математической корректности следовало бы учесть такую корреляцию, введя весовую матрицу, содержащую ковариации переменных. Тем не менее корреляцией можно пренебречь, так как спектрофотометрические ошибки начинают проявляться, когда ошибки в концентрациях составляют несколько десятых долей процента, а ошибки в измерении светопоглощения— несколько тысячных [12]. Показано [12], что даже в случае преобладания концентрационных ошибок пренебрежение корреляцией незначительно влияет на результат. [c.96]
В настоящее время метод остановленной струи широко приме-ляется для решения многих задач химической кинетики установление механизмов химической реакции, определение стадий, лимитирующих протекание реакции обнаружение промежуточных комплексов, определение кинетики ферментативных реакций, установление числа и концентрации активных центров фермента, изучение быстрых конформационны5( переходов в белках и нуклеиновых кислотах. Метод требует быстрой регистрации это единственное существенное ограничение его применимости. Особое внимание при применении метода остановленной струи необходимо уделять тер-мостатированию, так как разница в температурах в кювете наблюдения и растворе смеси реагентов может привести к большим оптическим ошибкам, затрудняющим установление механизма наблюдаемой реакции. Точность определения констант скоростей данным методом примерно такая, как и при обычных спектрофотометрических измерениях кинетики химических реакций. [c.28]
Рассмотрим в первую очередь ошибки, вытекающие из самой сущности законов поглощения излучений, и основные закономерности, установленные еще в 1937 г. Туайменом и Лотианом [19]. Найденная ими зависимость ошибки измерения А от ее абсолютного значения является определяющей в оценке ошибок спектрофотометрических измерений. [c.30]
Иногда в случае неблагоприятных условий спектрофотометрические измерения подвержены более высоким ошибкам. Например, реакцию гафния (IV) с хлораниловой кислотой в 3 М хлорной кислоте изучали спектрофотометрически в области 260—360 нм. Было измерено светопоглощение двенадцати растворов при двадцати одной длине волны [6]. На рис. 2.4 показана зависимость определяемого числа частиц от заданной ошибки матричного элемента. Результаты, полученные при исследовании хлораниловой кислоты в 3 М хлорной кислоте, также показаны на рис. 2.4. [c.41]
Определение по собственному светопоглощению. Метод основан на спектрофотометрическом измерении светопоглощения водного, раствора хлора [164, 524] или его раствора в I4 [117, 946] в УФ-области спектра (330—350 нм). Нижний предел определяемой концентрации хлора 2-10 М (1 мкг мл). Относительная ошибка при определении 10 М хлора составляет 4%, для более низких концентраций (< 10 М) ошибка увеличивается до 30— 50% [117]. [c.68]
Для индикации и регистрации показаний анализаторов все шире применяют цифровые преобразователи и цифровые регистраторы, обладающие целым рядом преимуществ перед аналоговыми, к которым в первую очередь относятся почти полное устранение ошибки считывания показаний и возможность непосредственной обработки данных на вычислительной машине. В состав цифрового регистрирующего устройства входят аналого-цифровой преобразователь, индикатор и перфоратор с логической схемой управления. Голландской фирмой Витатрон для регистрации и обработки результатов спектрофотометрических измерений выпускаются цифровой преобразователь ОНР 100 и цифровой регистратор ОКР 200. [c.135]
Спектрофотометрические измерения. На рис. 1 показаны ультрафиолетовые спектры перксеноната при различных значениях pH. Поскольку ионную силу растворов не контролировали, о результатах спектрометрических измерений можно сделать только качественные заключения. Однако очевидно, что при pH ниже И в перксенонатном растворе появляются новые формы соединений ксенона. В пределах ошибки опыта имеются две изобестических точки, что указывает на то, что во всем интервале pH имеются только две основные поглощающие формы. [c.237]
Свегла, Палл и Ердеи показали , что для вычисления ошибок спектрофотометрических измерений недостаточно учитывать только ошибки показания прибора ошибки возникают также из-за неопределенности отрезков, отсекаемых на осях координат калибровочными графиками и из-за различных наклонов последних. [c.30]
Сопоставление активационных параметров кислотного гидролиза алкилсульфатов с длинной цепью и немицеллярного этилсуль-фата показывает, что ускорение реакции при образовании мицелл связано главным образом с уменьшением энтальпии активации, а не с увеличением энтропии [212]. Этот вывод был получен с использованием потенциометрических данных. Однако энергия активации кислотного гидролиза додецилсульфата натрия, полученная из спектрофотометрических измерений, оказалась одинаковой в мицеллярных и истинных растворах (табл. 8), тогда как энтропия активации была на 6,9 энтр. ед. больше в случае мицеллярного раствора [215]. Это противоречие, вероятно, объясняется неодинаковым выбором стандарта для сравнения (раствор этилсульфата и неми-целлярный раствор додецилсульфата). Возможно также, что расхождения связаны с отклонениями температурной зависимости от уравнения Аррениуса и зависящими от температуры ошибками потенциометрического метода. [c.282]
Такой спектральный анализ требует трудно достижимой точности спектрофотометрических измерений. Он осложнен взаимным наложением аналитических полос свободных молекул и Н-комплексов, температурной зависимостью коэффициентов поглощения этих полос, неудобством и неточностью термостатирования (из-за нагрева образца излучением) и другими факторами. Ошибки спектральных определений АН составляют от 0,2 ккал1моль для прецизионных измерений до 0,5—1 ккалЫоль для обычных, но эти величины скорее характеризуют лишь невоспроизводимость измерений, а действительные неточности больше Расхождения результатов разных авторов часто намного превышает предполагаемые ошибки, достигая целых единиц ккалЫоль, или приблизительно 100% от измеряемой величины Поэтому, несмотря на большое число опубликованных данных, надежные сходящиеся значения энтальпий водородных связей известны все еще для сравнительно немногих систем . [c.139]
В работе [1049] изучены условия, при которых возможно быстрое спектрофотометрическое определение ртути в неорганических соединениях. Показано, что закон Вера выполняется для концентраций (0,5—4)-10 М Hg(II). Относительное стандартное отклонение составляет 1,8%. Изучено влияние концентрации иодида калия на определение ртути и найдено, что для 2,2-10 М Hg(II) поглощение остается неизменным, если концентрация иодида калия изменяется от 1,2 до 0,8 М. Установлено, что при pH 4 окисление Т до Тз становится заметным, однако ошибка не превышает 1%. Измерение поглощения ртутного комплекса при pH 10 дает ошибку 1%. Низкие величины оптической плотности могут быть получены при высоких pH из-за образования частиц Hg(OH) . На определение ртути данным методом оказывают влияние анионы СгО , СгзО , поглощающие в области 323 млг. Влияние СН связано с образованием частиц типа Hg( N) J4 . Ионы Ag , Сг + не влияют, если их концентрация равна 2-10 М. Но медь, платина, золото окисляют Т до и поэтому должны быть восстановлены кислым раствором НааЗгОз до анализа. Влияют на определение ртути ионы Ре(П), РЬ(П), В1(1П), Т1(1), которые дают видимые осадки в 1 М КТ при концентрации их. <1.10 М. Этот метод может быть применен в присутствии галогенидов и псевдогалогенидов. [c.105]
Одна из групп исследователей [16] вычисляла константы устойчивости, используя уравнения материального баланса. Минимизировалась сумма квадратов отклонений аналитической концентрации иона водорода. В этом случае взвешивание особенно важно, поскольку ошибка измерения pH соответствует большим отклонениям при низких значениях pH, чем при высоких [13]. Обычно взвешивание более необходимо при потенциометрических вычислениях, чем в спектрофотометрических методах 1 жно оно и тогда, когда используются отклонения функции п. Оказалось, что вычисленные веса изменяются в слишком широких пределах [26, 68, 69]. Возможно, частичной причиной этого является то, что авторы аппроксимируют данные функцией, зависимые переменные которой сами являются функциями экспериментальных наблюдений. Так, очевидно, что полная аналитическая концентрация иона водорода является экспоненциальной функцией от pH. Таким образом, условия применимости метода наименьших квадратов (разд. 4.6) выполнены не полностью, поскольку неточные зависимые переменные сопоставляются с функциями от точных значений независимых переменных. Особенно следует избегать использования отклонений функции образования п. Правильным будет применять для расчета всех потенциометрических данных функцию суммы квадратов разностей между вычисленными и наблюдаемыми э. д. с. Дополнительное преимущество такого подхода — возможность использовать единичные веса до тех пор, пока нет веских оснований полагать противное. Примером использования единичных весов служит минимизация суммы квадратов разностей меладу вычисленным и наблюдаемым объемом титрантов в процессе кислотно-основного титрования [29]. Другие исследователи также для простоты вводили допущение о единичности весовой матрицы [11, 15, 31, 51], и было сообщение, что и с весовыми коэффициентами и без них получались одни и те же значения рассчитанных констант устойчивости. [c.95]
Спектрофотометрия в ультрафиолетовой (УФ) и видимой областях (ОФС.1.2.1.1.0003.15)
Государственная фармакопея 13 издание (ГФ XIII)
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
Взамен ОФС ГФ X, ОФС ГФ XI, ОФС 42-0042-07 ГФ XII, ч.1
Спектроскопические методы анализа основаны на избирательном поглощении электромагнитного излучения анализируемым веществом и служат для исследования строения, идентификации и количественного определения светопоглощающих соединений.
В зависимости or используемой аппаратуры в фармацевтическом анализе различают следующие методы анализа, основанные на поглощении электромагнитного излучения и испускании света:
- спектрофотометрия в ультрафиолетовой (УФ) и видимой областях;
- спектрометрия в инфракрасной (ИК) области;
- атомно-эмиссионная спектрометрия (АЭС);
- атомно-абсорбционная спектроскопия (ААС);
- флуоримегрия;
- спектроскопия ядерного магнитного резонанса (ЯМР);
- масс-спектрометрия;
- рамановская спектрометрия;
- рентгеновская флуоресцентная спектрометрия;
- рентгеновская порошковая дифрактометрия.
Ряд длин волн, для которых проводятся измерения методами абсорбционной спектрофотометрии, охватывает спектральную область от коротких длин волн в УФ-области до ИК-области. Для удобства отнесений этот спектральный ряд делится на следующие диапазоны длин волн: УФ (от 190 до 380 нм), видимый (от 380 до 780 нм), ИК (от 0,78 до 400 мкм).
СПЕКТРОФОТОМЕТРИЯ В УЛЬТРАФИОЛЕТОВОЙ И ВИДИМОЙ ОБЛАСТЯХ
Уменьшение интенсивности монохроматического излучения, проходящего через гомогенную поглощающую среду, количественно описывается законом Бугера-Ламберта-Бера:
![]()
где:
Т – пропускание, отношение интенсивности светового потока, прошедшего через вещество, к интенсивности падающего на вещество светового потока: Т = I/I0;
I – интенсивность прошедшего монохроматического излучения;
I0 – интенсивность падающего монохроматического излучения;
ε – молярный показатель поглощения;
с – молярная концентрация вещества в растворе;
b – длина оптического пути или толщина слоя, в сантиметрах.
Величина log10(1/Т) носит название оптической плотности, обозначается буквой А и является измеряемой величиной. В отсутствии других физико-химических факторов измеренная оптическая плотность (А) пропорциональна концентрации вещества в растворе (с) и толщине слоя (b).
Величина ![]() представляет собой удельный показатель поглощения, т.е. оптическую плотность раствора вещества с концентрацией 10 г/л (1 г/100 мл) в кювете с толщиной слоя 1 см. Величины
представляет собой удельный показатель поглощения, т.е. оптическую плотность раствора вещества с концентрацией 10 г/л (1 г/100 мл) в кювете с толщиной слоя 1 см. Величины![]() и ε связаны соотношением:
и ε связаны соотношением:
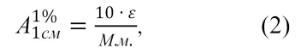
где:
М.м. – молекулярная масса исследуемого вещества.
Измерение оптической плотности
Если нет других указаний в фармакопейной статье, измерение оптической плотности проводят при указанной длине волны с использованием кювет с толщиной слоя 1 см и при температуре (20 ± 1) °С по сравнению с тем же растворителем или той же смесью растворителей, в которой растворено вещество. При измерении оптической плотности раствора при данной длине волны оптическая плотность кюветы с растворителем, измеренная против воздуха при той же длине волны, не должна превышать 0,9 и, желательно, чтобы она была не менее 0,2.
Спектр поглощения представляют таким образом, чтобы оптическая плотность или ее некоторая функция были приведены по оси ординат, а длина волны или некоторая функция длины волны – по оси абсцисс.
Если в фармакопейной статье для максимума поглощения указывается только одна длина волны, то это означает, что полученное значение максимума не должно отличаться от указанного более чем на ± 2 нм.
Приборы
Спектрофотометры, предназначенные для измерений в ультрафиолетовой и видимой областях спектра, состоят из оптической системы, выделяющей монохроматическое излучение в области от 190 до 800 нм и обеспечивающей его прохождение через образец, и устройства для измерения оптической плотности.
Основными частями этих приборов являются: источник излучения, диспергирующий прибор (призма или решетка), щель для выделения полосы длин волн, кюветы для образцов, детектор излучаемой энергии, встроенные усилители и измерительные приборы.
Проверка шкалы длин волн в ультрафиолетовой и видимой области. Точность калибровки прибора по шкале длин волн в спектральном ряду проверяют по приведенным в табл. 1 спектральным линиям водородной (Hβ) или дейтериевой (Dβ) разрядной лампы, линиям паров ртути (Hg) кварцево-ртутной дуговой лампы, а также по максимумам поглощения раствора гольмия перхлората (Ho) (готовый реактив для калибровки спектрофотометра представляет собой 4 % раствор гольмия оксида в 14,1% растворе хлорной кислоты). Допустимое отклонение составляет ± 1 нм для ультрафиолетовой и ± 3 нм для видимой области.
Таблица 1. Максимумы поглощения для проверки шкалы длин волн
| 241,15 нм (Но) | 404,66 нм (Hg) |
| 253,7 нм (Hg) | 435,83 нм (Hg) |
| 287,15 нм (Но) | 486,0 нм (Dв) |
| 302,25 нм (Hg) | 486,1 нм (Нв) |
| 313,16 нм (Hg) | 536,3 нм (Но) |
| 334,15 нм (Hg) | 546,07 нм (Hg) |
| 361,5 нм (Но) | 576,96 нм (Hg) |
| З65,48 нм (Hg) | 579,07 нм (Hg) |
Шкала длин волн может быть калибрована также при помощи подходящих стеклянных фильтров, которые имеют фиксированные полосы поглощения в видимой и ультрафиолетовой областях, а также стандартных стекол, содержащих дидим (смесь празеодима и неодима), и стекол, содержащих гольмий.
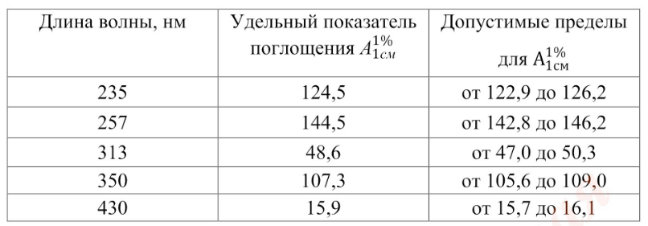
Проверка шкалы оптической плотности. Для проверки шкалы оптической плотности используют стандартные неорганические стеклянные фильтры или раствор калия дихромата при длинах волн, указанных в табл. 2, где для каждой длины волны приведено точное значение удельного показателя поглощения ![]() и допустимые пределы.
и допустимые пределы.
Раствор калия дихромата для проверки шкалы оптической плотности при 235, 257, 313 и 350 нм готовят следующим образом: от 57,0 до 63,0 мг (точная навеска) калия дихромата, предварительно высушенного до постоянной массы при температуре 130 °С, растворяют в 0,005 М растворе серной кислоты и доводят объем раствора тем же растворителем до 1000 мл. Для проверки оптической плотности при 430 нм, растворяют 57,0-63,0 мг (точная навеска) калия дихромата в 0,005 М растворе серной кислоты и доводят объём раствора тем же растворителем до метки.
Таблица 2. Удельный показатель поглощения стандартов при различных длинах волн
Предельный уровень рассеянного света. Рассеянный свет может быть обнаружен при данной длине волны с использованием соответствующих фильтров или растворов: например, оптическая плотность раствора 12 г/л калия хлорида в кювете с толщиной слоя 1 см резко увеличивается между 220 и 200 нм и должна быть больше 2 при 198 нм при использовании воды в качестве раствора сравнения.
Разрешающая способность (для качественного анализа). Если есть указание в фармакопейной статье, определяют разрешающую способность спектрофотометра следующим образом. Записывают спектр 0,02 % (об/об) раствора толуола в гексане. Минимально допустимое значение отношения оптической плотности в максимуме поглощения при 269 нм к оптической плотности в минимуме поглощения при 266 нм указывают в фармакопейной статье.
Ширина спектральной щели (для количественного анализа). В случае использования спектрофотометра с изменяемой шириной спектральной щели при выбранной длине волны возможны погрешности, связанные с шириной этой щели. Для их исключения ширина щели должна быть малой по сравнению с полушириной полосы поглощения (шириной на половине оптической плотности) и в то же время должна быть максимально велика для получения высокого значения интенсивности падающего монохроматического излучения (I0). Таким образом, ширина щели должна быть такой, чтобы дальнейшее ее уменьшение не изменяло величину измеряемой оптической плотности.
Кюветы. Допустимые отклонения в толщине слоя используемых кювет должны быть не более ±0,005 см. Кюветы, предназначенные для испытуемого раствора и раствора сравнения, должны иметь одинаковое пропускание (или оптическую плотность) при заполнении одним и тем же растворителем. В противном случае это различие следует учитывать.
Требования к растворителям. Для определений, производимых в ультрафиолетовой и видимой областях, образец анализируемого вещества растворяют в соответствующем растворителе, который должен быть оптически прозрачным в используемой области длин волн. Для этих областей длин волн пригодны многие растворители, в том числе вода, спирты, хлороформ, низшие углеводороды, эфиры и разбавленные растворы сильных кислот и щелочей.
Идентификация
Абсорбционную спектрофотометрию в ультрафиолетовой и видимой областях спектра применяют для определения подлинности лекарственных средств путем:
- сравнения спектров поглощения испытуемого раствора и раствора стандартного образца; в указанной области спектра должно наблюдаться совпадение положений максимумов, минимумов, плеч и точек перегиба;
- указания положений максимумов, минимумов, плеч и точек перегиба спектра поглощения испытуемого раствора; расхождение между наблюдаемыми и указанными длинами волн в максимумах и минимумах поглощения не должно обычно превышать ± 2 нм.
Возможны и другие варианты применения, оговоренные в фармакопейных статьях.
Количественное определение
Определение концентрации веществ спектрофотометрическим методом основано на использовании закона Бугера-Ламберта-Бера:
В ряде случаев, даже при использовании монохроматического излучения могут наблюдаться отклонения от закона Бугера-Ламберта-Бера, обусловленные процессами диссоциации, ассоциации и комплексообразования. Поэтому предварительно следует проверить линейность зависимости оптической плотности раствора от концентрации в аналитической области. При наличии отклонений от линейной зависимости следует пользоваться не формулой (3), а экспериментально найденной зависимостью.

Обычно определение концентрации спектрофотометрическим методом проводят с использованием стандартного образца. Расчет концентрации основан на использовании уравнения:
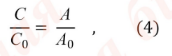 где:
где:
С и С0 – концентрации испытуемого раствора и раствора стандартного образца, соответственно;
А и А0 – оптические плотности испытуемого раствора и раствора стандартного образца, соответственно.
Концентрации испытуемого и стандартного раствора должны быть близки.
Вначале измеряют оптическую плотность раствора стандартного образца, приготовленного, как указано в фармакопейной статье, затем проводят измерение оптической плотности испытуемого раствора. Второе измерение проводят сразу после первого, с использованием той же кюветы, в тех же экспериментальных условиях.
Метод с использованием стандартного образца является более точным и надежным. Возможность применения значения удельного показателя поглощения в каждом конкретном случае следует обосновывать. Обычно метод с использованием значения удельного показателя поглощения применим при допусках содержания анализируемого вещества не менее ±10 % от номинального содержания.
Многокомпонентный спектрофотометрический анализ
Многокомпонентный спектрофотометрический анализ (анализ смесей) применяют для одновременного количественного определения нескольких компонентов лекарственных средств, каждое из которых подчиняется закону Бугера-Ламберта-Бера.
Количественное определение в многокомпонентном спектрофотометрическом анализе основывается обычно на использовании уравнения:
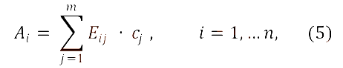 где:
где:
Аi – оптическая плотность испытуемого раствора при i-ой длине волны;
Еij – показатели поглощения (зависящие от способа выражения концентрации) j-го компонента образца при i-ой аналитической длине волны;
cj – концентрация j-го компонента образца.
Соответствующие методики проведения анализа и расчетные формулы указываются в фармакопейных статьях.
Производная спектрофотометрия
В производной спектрофотометрии исходные спектры поглощения (нулевого порядка) преобразуются в спектры производных первого, второго и более высокого порядков.
Спектр первой производной представляет собой график зависимости градиента кривой поглощения (скорость изменения оптической плотности от длины волны, dA/dλ) от длины волны.
Спектр второй производной представляет собой график зависимости кривизны спектра поглощения (d2A/dλ2) от длины волны. Вторая производная при любой длине волны связана с концентрацией следующим соотношением:

Производная спектрофотометрия может быть использована как для целей идентификации веществ, так и для их количественного определения в многокомпонентных смесях, а также в тех случаях, когда имеется фоновое поглощение, вызванное присутствием веществ, содержание которых не регламентируется.
Приборы
Используют спектрофотометры, отвечающие указанным выше требованиям и оснащенные аналоговым резистивно-емкостным дифференцирующим модулем или цифровым дифференциатором, или другими средствами получения производных спектров, в соответствии с инструкцией к прибору. Некоторые методы получения спектров второй производной приводят к смещению длин волн относительно исходного спектра, что следует учитывать там, где это необходимо.
Разрешающая способность
Если указано в фармакопейных статьях, записывают спектр второй производной для раствора 0,2 г/л толуола в метаноле, используя метанол в качестве раствора сравнения. На спектре должен присутствовать небольшой отрицательный экстремум, расположенный между двумя большими отрицательными экстремумами при 261 нм и 268 нм, в соответствии с рис. 1. Если нет других указаний в фармакопейных статьях, отношение А/B должно быть не менее 0,2.
Методика
Процедура анализа аналогична применяемой в обычной спектрофотометрии, но вместо оптических плотностей используют производные. Готовят раствор испытуемого образца, настраивают прибор в соответствии с инструкцией производителя и рассчитывают количество определяемого вещества, как указано в фармакопейной статье.

Рисунок 1. Спектр второй производной раствора толуола (0,2 г/л) в метаноле
Оптическая плотность раствора
Колориметрия
Из оптических методов анализа в практике аналитических лабораторий наиболее широко применяются колориметрические методы (от лат. color — цвет и греч. μετρεω — измеряю). Колориметрические методы основаны на измерении интенсивности светового потока, прошедшего через окрашенный раствор.
В колориметрическом методе используются химические реакции, сопровождающиеся изменением цвета анализируемого раствора. Измеряя светопоглощение такого окрашенного раствора или сравнивая полученную окраску с окраской раствора известной концентрации, определяют содержание окрашенного вещества в испытуемом растворе.
Существует зависимость между интенсивностью окраски раствора и содержанием в этом растворе окрашенного вещества. Эта зависимость, называемая основным законом светопоглощения (или законом Бугера—Ламберта—Бера), выражается уравнением:
I = I0 10 — ε c l
где I — интенсивность света, прошедшего через раствор; I0 — интенсивность падающего на раствор света; ε- коэффициент светопоглощения, постоянная величина для каждого окрашенного вещества, зависящая от его природы; С — молярная концентрация окрашенного вещества в растворе; l — толщина слоя светопоглощающего раствора, см.
Физический смысл этого закона можно выразить следующим образом. Растворы одного и того же окрашенного вещества при одинаковой концентрации этого вещества и толщине слоя раствора поглощают равное количество световой энергии, т. е. светопоглощение таких растворов одинаковое.
Для окрашенного раствора, заключенного в стеклянную кювету с параллельными стенками, можно сказать, что по мере увеличения концентрации и толщины слоя раствора его окраска увеличивается, а интенсивность света I, прошедшего через поглощающий раствор, уменьшается по сравнению с интенсивностью падающего света I0.
Рис.1 Прохождение света через кювету с исследуемым раствором.
Оптическая плотность раствора.
Если прологарифмировать уравнение основного закона светопоглощения и изменить знаки на обратные, то уравнение принимает вид:
Величина является очень важной характеристикой окрашенного раствора; ее называют оптической плотностью раствора и обозначают буквой A:
A = ε C l
Из этого уравнения вытекает, что оптическая плотность раствора прямо пропорциональна концентрации окрашенного вещества и толщине слоя раствора.
Другими словами, при одинаковой толщине слоя раствора данного вещества оптическая плотность этого раствора будет тем больше, чем больше в нем содержится окрашенного вещества. Или, наоборот, при одной и той же концентрации данного окрашенного вещества оптическая плотность раствора зависит только от толщины его слоя. Отсюда может быть сделан следующий вывод: если два раствора одного и того же окрашенного вещества имеют различную концентрацию, одинаковая интенсивность окраски этих растворов будет достигнута при толщинах их слоев, обратно пропорциональных концентрациям растворов. Этот вывод очень важен, так как на нем основаны некоторые методы колориметрического анализа.
Таким образом, чтобы определить концентрацию (С) окрашенного раствора, необходимо измерить его оптическую плотность (A). Чтобы измерить оптическую плотность, следует измерить интенсивность светового потока.
Интенсивность окраски растворов можно измерять различными методами. Различают субъективные (или визуальные) методы колориметрии и объективные (или фотоколориметрические).
Визуальными называются такие методы, при которых оценку интенсивности окраски испытуемого раствора делают невооруженным глазом.
При объективных методах колориметрического определения для измерения интенсивности окраски испытуемого раствора вместо непосредственного наблюдения пользуются фотоэлементами. Определение в этом случае проводят в специальных приборах — фотоколориметрах, откуда и метод получил название фотоколориметрического.
Визуальные методы
К визуальным методам относятся:
1) метод стандартных серий;
2) метод дублирования (колориметрическое титрование);
3) метод уравнивания.
Метод стандартных серий. При выполнении анализа методом стандартных серий интенсивность окраски анализируемого окрашенного раствора сравнивают с окрасками серии специально приготовленных стандартных растворов (при одинаковой толщине поглощающего слоя).
Растворы в колориметрии обычно имеют интенсивную окраску, поэтому имеется возможность определять весьма небольшие концентрации или количества веществ. Однако это может сопровождаться определенными трудностями: так навески для приготовления серии стандартных растворов могут быть очень малы. Для преодоления этих трудностей готовят стандартный раствор А достаточно высокой концентрации, например 1 мг/мл. После этого путем разбавления из раствора А готовят стандартный раствор В значительно меньшей концентрации, а из него в свою очередь готовят серию стандартных растворов.
Для этого в пробирки или кюветы одинакового размера и одинакового цвета стекла пипеткой добавляются необходимые объемы растворов реагентов в нужной последовательности. Порции растворов определяемого вещества целесообразно добавлять из бюретки, т.к. их объемы будут различны для обеспечения различных концентраций в серии стандартных растворов. При этом начальный раствор должен содержать все компоненты, кроме определяемого вещества (нулевой раствор). В исследуемый раствор добавляют растворы необходимых реагентов. Все растворы доводят до постоянного объема, а затем визуально сравнивают интенсивность окраски исследуемого раствора с растворами серии стандартных растворов. Возможно совпадение интенсивности окраски с каким-либо раствором серии. Тогда считается, сто исследуемый раствор имеет такую же концентрацию или содержит столько же определяемого вещества. Если же интенсивность окраски покажется промежуточной между соседними растворами серии, концентрация или содержание определяемого компонента считают средним арифметическим между растворами серии.
Колориметрическое титрование (метод дублирования). Этот метод основан на сравнении окраски анализируемого раствора с окраской другого раствора— контрольного. Для приготовления контрольного раствора готовят раствор, содержащий все компоненты исследуемого раствора, за исключением определяемого вещества, и все употреблявшиеся при подготовке пробы реактивы, и к нему добавляют из бюретки стандартный раствор определяемого вещества. Когда этого раствора будет добавлено столько, что интенсивности окраски контрольного и анализируемого раствора уравняются, считают, что в анализируемом растворе содержится столько же определяемого вещества, сколько его было введено в контрольный раствор.
Метод уравнивания.Этот метод основан на уравнивании окрасок анализируемого раствора и раствора с известной концентрацией определяемого вещества — стандартного раствора. Существуют два варианта выполнения колориметрического определения этим методом.
По первому варианту уравнивание окрасок двух растворов с разной концентрацией окрашенного вещества проводят путем изменения толщины слоев этих растворов при одинаковой силе проходящего через растворы светового потока. При этом, несмотря на различие концентраций анализируемого и стандартного растворов, интенсивность светового потока, проходящего через оба слоя этих растворов, будет одинакова. Соотношение между толщинами слоев и концентрациями окрашенного вещества в растворах в момент уравнивания окрасок будет выражаться уравнением:
где l1 — толщина слоя раствора с концентрацией окрашенного вещества C1, а l2-толщина слоя раствора с концентрацией окрашенного вещества C2.
В момент равенства окрасок отношение толщин слоев двух сравниваемых растворов обратно пропорционально отношению их концентраций.
На основании приведенного уравнения, измерив толщину слоев двух одинаково окрашенных растворов и зная концентрацию одного из этих растворов, легко можно рассчитать неизвестную концентрацию окрашенного вещества в другом растворе.
Для измерения толщины слоя, через который проходит световой поток, можно применять стеклянные цилиндры или пробирки, а при более точных определениях специальные приборы — колориметры.
По второму варианту, для уравнивания окрасок двух растворов с различной концентрацией окрашенного вещества, через слои растворов одинаковой толщины пропускают световые потоки различной интенсивности.
В этом случае оба раствора имеют одинаковую окраску, когда отношение логарифмов интенсивностей падающих световых потоков равно отношению концентраций.
В момент достижения одинаковой окраски двух сравниваемых растворов, при равной толщине их слоев, концентрации растворов прямо пропорциональны логарифмам интенсивностей падающего на них света.
По второму варианту определение может быть выполнено только с помощью колориметра.
Теоретические основы определения оптической плотности раствора
Любая частица, будь то молекула, атом или ион, в результате поглощения кванта света переходит на более высокий уровень энергетического состояния. Чаще всего осуществляется переход из основного в возбужденное состояние. Это вызывает появление в спектрах определенных полос поглощения.
Поглощение излучения приводит к тому, что при пропускании его через вещество интенсивность этого излучения снижается при увеличении количества частиц вещества, обладающего некоторой оптической плотностью. Этот метод исследования предложил В. М. Севергин еще в 1795 году.
Наилучшим образом этот метод годится для реакций, где определяемое вещество способно переходить в окрашенное соединение, что вызывает изменение окраски исследуемого раствора. Измерив его светопоглощение или сравнив окраску с раствором известной концентрации, несложно найти процент содержания вещества в растворе.
Основной закон светопоглощения
Суть фотометрического определения заключается в двух процессах:
- перевод определяемого вещества в поглощающее электромагнитные колебания соединение;
- замер интенсивности поглощения этих самых колебаний раствором исследуемого вещества.
Изменения в интенсивности потока света, проходящего через светопоглощающее вещество, будут вызываться также потерями света из-за отражения и рассеяния. Чтобы результат был достоверным, проводят параллельные исследования по замеру параметров при той же толщине слоя, в идентичных кюветах, с тем же растворителем. Так снижение интенсивности света зависит главным образом от концентрации раствора.
Уменьшение интенсивности света, пропущенного через раствор, характеризуют коэффициентом светопропускания (также принято называть его пропусканием) Т:
- I — интенсивность света, пропущенного через вещество;
- I0 — интенсивность падающего пучка света.
Таким образом, пропускание показывает долю непоглощенного светового потока, проходящего через изучаемый раствор. Обратный алгоритм значения пропускания называют оптической плотностью раствора (D): D = (-lgT) = (-lg) * (I / I0) = lg * (I0 / I).
Это уравнение показывает, какие параметры являются главными для исследования. К ним относится длина волны света, толщина кюветы, концентрация раствора и оптическая плотность.
Закон Бугера-Ламберта-Бера
Он является математическим выражением, отображающим зависимость уменьшения интенсивности монохроматического потока света от концентрации светопоглощающего вещества и толщины жидкостного слоя, через который он пропущен:
I = I0 * 10 -ε·С·ι , где:
- ε — коэффициент поглощения света;
- С — концентрация вещества, моль/л;
- ι —толщина слоя анализируемого раствора, см.
Преобразовав, эту формулу можно записать: I / I0 = 10 -ε·С·ι .
Суть закона сводится к следующему: различные растворы одного и того же соединения при равной концентрации и толщине слоя в кювете поглощают одинаковую часть падающего на них света.
Прологарифмировав последнее уравнение, можно получить формулу: D = ε * С * ι.
Очевидно, что оптическая плотность напрямую зависит от концентрированности раствора и толщины его слоя. Становится ясен физический смысл молярного коэффициента поглощения. Он равен D для одномолярного раствора и при толщине слоя в 1 см.
Ограничения применения закона
Этот раздел включает следующие пункты:
- Он справедлив исключительно для монохроматического света.
- Коэффициент ε связан с показателем преломления среды, особенно сильные отклонения от закона могут наблюдаться при анализе высококонцентрированных растворов.
- Температура при измерении оптической плотности должна быть постоянной (в рамках нескольких градусов).
- Световой пучок должен быть параллельным.
- рН среды должен быть постоянным.
- Закон применим для веществ, светопоглощающими центрами которых являются частицы одного вида.
Методы определения концентрации
Стоит рассмотреть метод градуировочного графика. Для его построения готовят ряд растворов (5-10) с различной концентрацией исследуемого вещества и замеряют их оптическую плотность. По полученным значениям выстраивают график зависимости D от концентрации. График является прямой линией, идущей от начала координат. Он позволяет легко определить концентрацию вещества по результатам проведенных измерений.
Также существует метод добавок. Применяется реже, чем предыдущий, но позволяет проанализировать растворы сложного состава, поскольку учитывает влияние дополнительных компонентов. Суть его состоит в определении оптической плотности среды Dx, содержащей определяемое вещество неизвестной концентрации Сх, с повторным анализом того же раствора, но с добавлением определенного количества исследуемого компонента (Сст). Величину Сх находят, используя расчеты или графики.
Условия проведения исследования
Чтобы фотометрические исследования давали достоверный результат, необходимо соблюдать несколько условий:
- реакция должна заканчиваться быстро и полностью, избирательно и воспроизводимо;
- окраска образующегося вещества должна быть устойчива во времени и не изменяться под действием света;
- исследуемое вещество берут в количестве, которого достаточно для перевода его в аналитическую форму;
- замеры оптической плотности проводят в том интервале длин волн, при котором различие в поглощении исходных реагентов и анализируемого раствора наибольшее;
- светопоглощение раствора сравнения принято считать оптическим нулем.
Способы расчета концентрации по величине аналитического сигнала
СПОСОБЫ РАСЧЕТА КОНЦЕНТРАЦИИ ПО ВЕЛИЧИНЕ АНАЛИТИЧЕСКОГО СИГНАЛА
МЕТОД ГРАДУИРОВОЧНОГО ГРАФИКА
Пример 1. При измерении оптической плотности в одинаковых условиях (длина волны 340 нм, толщина поглощающего слоя – 1,00 см) растворов калия дихромата с разной концентрацией хрома (мкг/мл) получены следующие результаты:
Изобразите примерный вид градуировочного графика; методом наименьших квадратов рассчитайте обратное уравнение градуировочного графика с = bA + а; определите концентрацию хрома (мкг/мл) в растворе Х, имеющем оптическую плотность 0,480.
Рассчитайте массу хрома (мг) в анализируемой пробе, если ее растворили в присутствии концентрированной серной кислоты в воде дистиллированной в мерной колбе объёмом 50,00 мл (раствор Х).
1. Расчет методом наименьших квадратов
2. Расчет с помощью программы Excel
длина волны 340 нм
Ответ: с = 115,72А — 8,8397 (r = 0,9941); масса хрома в пробе 2,34 мг
Пример 2. При измерении оптической плотности в одинаковых условиях (длина волны 400 нм, толщина поглощающего слоя – 1,00 см) растворов никеля (II) нитрата с разной концентрацией никеля (мг/мл) получены следующие результаты:
Изобразите примерный вид градуировочного графика; методом наименьших квадратов рассчитайте обратное уравнение градуировочного графика с = bA + а; определите концентрацию никеля (мг/мл) в растворе Х, имеющем оптическую плотность 0,350.
Рассчитайте массу никеля (мг) в анализируемой пробе, если ее количественно перенесли в мерную колбу объёмом 25,00 мл и развели водой дистиллированной в присутствии азотной кислоты до метки (раствор Х).
1. Расчет методом наименьших квадратов
2. Расчет с помощью программы Excel
Ответ: с = 42,495А — 3,8535 (r = 0,9986); масса никеля 275 мг
Пример 3. При измерении оптической плотности в одинаковых условиях (длина волны 620 нм, толщина поглощающего слоя – 1,00 см) растворов меди (II) в виде аммиачного комплекса с разной концентрацией меди (мкг/мл) получены следующие результаты:
Изобразите примерный вид градуировочного графика; методом наименьших квадратов рассчитайте обратное уравнение градуировочного графика с = bA; определите концентрацию меди (мкг/мл) в растворе Х, имеющем оптическую плотность 0,150.
Рассчитайте массу меди (мкг) в анализируемой пробе, если ее количественно перенесли в мерную колбу объёмом 25,00 мл и до метки развели водой дистиллированной в присутствии избытка аммиака (раствор Х).
1. Расчет методом наименьших квадратов
2. Расчет с помощью программы Excel
2. Расчет с помощью программы Excel
Ответ: с = 34,595А (r = 0,9985); масса меди 130 мкг.
МЕТОД ОДНОГО СТАНДАРТНОГО РАСТВОРА
Измеряют величину аналитического сигнала (yст) для раствора с известной концентрацией вещества (сст). Затем измеряют величину аналитического сигнала (yx) для раствора с неизвестной концентрацией вещества (сx). Такой способ расчёта можно использовать в том случае, если зависимость аналитического сигнала от концентрации описывается линейным уравнением без свободного члена. Концентрация вещества в стандартном растворе должна быть такой, чтобы величины аналитических сигналов, полученных при использовании стандартного раствора и раствора с неизвестной концентрацией вещества, были бы как можно ближе друг к другу.
ПРИМЕР 1. При фотометрическом определении концентрации нитрит-ионов с помощью реактива Грисса (раствора сульфаниловой кислоты и α-нафтиламина в разбавленной уксусной кислоте) было установлено, что раствор с концентрацией нитрит-ионов 2,00 мкг/мл имеет в соответствующих условиях оптическую плотность 0,300. Рассчитайте концентрацию нитрит-ионов в растворе (мкг/мл), оптическая плотность которого в таких же условиях равна 0,250. Зависимость оптической плотности от содержания аналита линейна и проходит через начало координат.
Ответ: 1,67 мкг/мл
МЕТОД ДВУХ СТАНДАРТНЫХ РАСТВОРОВ
(метод ограничивающих растворов)
Измеряют величины аналитических сигналов для стандартных растворов с двумя разными концентрациями вещества, одна из которых (с1) меньше предполагаемой неизвестной концентрации (сx), а вторая (с2) – больше. Его используют, если зависимость аналитического сигнала от концентрации описывается линейным уравнением, не проходящим через начало координат.
Пример 1. Раствор с концентрацией никеля (II) 12,00 мг/мл имеет оптическую плотность 0,350 нм, а с концентрацией 16,00 мг/мл – 0,440. Определите концентрацию никеля (мг/мл) в растворе с оптической плотностью 0,380 (все измерения проводились в одинаковых условиях: длина волны 400 нм, толщина поглощающего слоя – 1,00 см, раствор в азотной кислоте).
Ответ: 13,33 мг/мл
Используют при анализе сложных матриц, когда матричные компоненты оказывают влияний на величину аналитического сигнала и невозможно точно скопировать матричный состав образца, в случае линейной зависимости, проходящей через начало координат.
Вначале измеряют величину аналитического сигнала (yx) для пробы с неизвестной концентрацией вещества. Затем к данной пробе прибавляют некоторое точное количество определяемого вещества (стандарта) и снова измеряют величину аналитического сигнала (yдоб). Концентрацию определяемого компонента в анализируемой пробе (без учета разбавления) рассчитывают по формуле:
Для учета разбавления раствора используем формулу:
ПРИМЕР 1. Раствор с неизвестной концентрацией вещества имел оптическую плотность 0,300. К 5,00 мл такого раствора прибавили 2,00 мл раствора с концентрацией этого же вещества 40,0 мг/л. Оптическая плотность полученного раствора при измерении её в таких же условиях оказалась равна 0,500. Рассчитайте концентрацию вещества (мг/л) в исходном растворе.
1 способ: пропорционально
2 способ: преобразуем составленную пропорцию в приведенную ранее формулу
ПРИМЕР 2. Оптическая плотность раствора с неизвестным содержанием вещества равна 0,400. При добавлении к анализируемому раствору 10,0 мкг этого же вещества оптическая плотность увеличилась до 0,500. Рассчитайте массу определяемого вещества (мкг) в исходном растворе.
1 способ: пропорционально
2 способ: преобразуем составленную пропорцию в приведенную ранее формулу
источники:
http://fb.ru/article/378024/teoreticheskie-osnovyi-opredeleniya-opticheskoy-plotnosti-rastvora
http://pandia.ru/text/80/260/2737.php
Стр. 2
плотности воды при 20 град. С (в г/ куб. см с учетом плотности
воздуха); 0,0012 - плотность воздуха при 20 град. С и
барометрическом давлении 1011 гПа (760 мм рт. ст.).
Метод 2. Применяют в случае определения плотности жидкостей с
точностью до 0,01. Испытуемую жидкость помещают в цилиндр и при
температуре жидкости 20 град. С осторожно опускают в нее чистый
сухой ареометр, на шкале которого предусмотрена ожидаемая величина
плотности. Ареометр не выпускают из рук до тех пор, пока не станет
очевидным, что он плавает; при этом необходимо следить, чтобы
ареометр не касался стенок и дна цилиндра. Отсчет производят через
3-4 мин после погружения по делению на шкале ареометра,
соответствующему нижнему мениску жидкости (при отсчете глаз должен
быть на уровне мениска).
Примечания. 1. Определение плотности сильнолетучих веществ
ареометром не допускается.
2. В случае определения темноокрашенных жидкостей отсчет
производят по верхнему мениску.
Метод 3. Применяют для определения плотности твердых жиров и
воска. Точно взвешивают пустой пикнометр, затем взвешивают тот же
пикнометр, наполненный дистиллированной водой, температура которой
20 град. С. После этого воду удаляют и пикнометр высушивают. Все
операции проводят, соблюдая условия, указанные в методе 1.
В пикнометр вливают при помощи пипетки или небольшой воронки с
тонкооттянутым концом расплавленный жир или воск в таком
количестве, чтобы он занимал 1/3 - 1/2 объема пикнометра.
Пикнометр ставят на один час без пробки в горячую воду, затем
охлаждают до 20 град. С и взвешивают; доводят до метки
дистиллированной водой при 20 град. С, вытирают насухо и вновь
взвешивают. В обеих фазах и на поверхности их раздела не должно
быть пузырьков воздуха.
Величину плотности вычисляют по следующей формуле:
(m2 - m) 0,99703
"ро20" = -------------------- + 0,0012,
(m1 + m2) - (m + m3)
где m - масса пустого пикнометра в граммах; m1 - масса
пикнометра с дистиллированной водой в граммах; m2 - масса
пикнометра с жиром в граммах; m3 - масса пикнометра с жиром и
водой в граммах.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ СПИРТА
В ФАРМАЦЕВТИЧЕСКИХ ПРЕПАРАТАХ
В круглодонную колбу вместимостью 200-250 мл отмеривают точное
количество жидкости. При содержании спирта в жидкости до 20% для
определения берут 75 мл жидкости, если жидкость содержит от 20 до
50% - 50 мл, от 50% и выше - 25 мл; жидкость перед перегонкой
разбавляют водой до 75 мл.
Для равномерного кипения в колбу с жидкостью помещают
капилляры, пемзу или кусочки прокаленного фарфора. Если жидкость
при перегонке сильно пенится, то добавляют фосфорную или серную
кислоту (2-3 мл), хлорид кальция, парафин или воск (2-3 г).
Приемник (мерную колбу вместимостью 50 мл) помещают в сосуд с
холодной водой, собирают около 48 мл отгона, доводят его
температуру до 20 град. С и добавляют воды до метки. Отгон должен
быть прозрачным или слегка мутным.
Плотность отгона определяют пикнометром и по
алкоголеметрическим таблицам находят соответствующее содержание
спирта в процентах по объему <*>.
--------------------------------
<*> См. с. 303.
Содержание спирта в препарате (X) в процентах по объему
вычисляют по формуле:
50а
Х= ---
б
где 50 - объем отгона в миллилитрах; а - содержание спирта в
процентах по объему; б - объем исследуемого препарата, взятый для
отгона, в миллилитрах.
Если испытуемая жидкость содержит летучие вещества - эфир,
эфирные масла, хлороформ, камфору, летучие кислоты или основания,
свободный йод и др., ее предварительно обрабатывают.
При содержании в жидкости эфира, эфирных масел, хлороформа,
камфоры к ней добавляют в делительной воронке равный объем
насыщенного раствора натрия хлорида и такой же объем петролейного
эфира. Смесь взбалтывают в течение 3 мин. После разделения слоев
спиртоводный слой сливают в другую делительную воронку и
обрабатывают таким же образом половинным количеством петролейного
эфира. Спиртоводный слой сливают в колбу для отгона, а соединенные
эфирные жидкости взбалтывают с половинным количеством насыщенного
раствора натрия хлорида, потом присоединяют к жидкости,
находящейся в колбе для отгона.
Если жидкость содержит менее 30% спирта, то высаливание
производят не раствором, а 10 г сухого натрия хлорида.
При содержании летучих кислот их нейтрализуют раствором щелочи,
при содержании летучих оснований - фосфорной или серной кислотой.
Жидкости, содержащие свободный йод, перед дистилляцией
обрабатывают цинковой пылью или рассчитанным количеством сухого
натрия тиосульфата до обесцвечивания. Для связывания летучих
сернистых соединений прибавляют несколько капель раствора едкого
натра.
Определение содержания спирта в настойках проводят также по
температуре кипения.
Прибор для количественного определения спирта в настойках
состоит из сосуда для кипячения 1, трубки 2 с боковым отростком,
холодильника 3 и ртутного термометра 4 с ценой деления 0,1 град. С
и пределом шкалы от 50 до 100 град. С (рис. 4) <*>.
--------------------------------
<*> Рис. 4. Прибор для количественного определения спирта в
настойках (объяснение в тексте). (Рисунок не приводится).
В сосуд для кипячения наливают 40 мл настойки и для
равномерного кипения помещают капилляры, пемзу или кусочки
прокаленного фарфора. Термометр помещают в приборе таким образом,
чтобы ртутный шарик выступал над уровнем жидкости на 2-3 мм.
Нагревают на сетке с помощью электроплитки мощностью 200 Вт или
газовой горелки. Когда жидкость в колбе начнет закипать, с помощью
реостата в 2 раза уменьшают напряжение, подаваемое на плитку.
Через 5 мин после начала кипения, когда температура становится
постоянной или ее отклонение не превышает +/- 0,1 град. С, снимают
показания термометра. Полученный результат приводят к нормальному
давлению. Если показания барометра отличаются от 1011 гПа (760 мм
рт. ст.), вносят поправку на разность между наблюдаемым и
нормальным давлением 0,04 град. С на 1,3 гПа (1 мм рт. ст.). При
давлении ниже 1011 гПа поправку прибавляют к установленной
температуре, при давлении выше 1011 гПа поправку вычитают.
Содержание спирта в настойке определяют при помощи таблицы.
Пример. Температура кипения настойки пустырника 80,9 град. С,
атмосферное давление 1000 гПа (752 мм рт. ст.), разность давлений
1011 - 1000 = 11 гПа (760 - 752 = 8 мм рт. ст.). Поправка
составляет: 0,04 град. С х 8 = 0,32 град. С. К найденной
температуре кипения прибавляют поправку: (80,9 + 0,32) град. С. По
таблице этой температуре кипения соответствует 66% спирта.
Определение концентрации спирта в водно - спиртовых смесях
по температуре кипения при давлении 1011 гПа
(760 мм рт. ст.)
------------T---------T-----------T---------T-----------T---------¬
¦Температура¦% спирта ¦Температура¦% спирта ¦Температура¦% спирта ¦
¦ кипения, ¦по объему¦ кипения, ¦по объему¦ кипения, ¦по объему¦
¦ град. С ¦ ¦ град. С ¦ ¦ град. С ¦ ¦
+-----------+---------+-----------+---------+-----------+---------+
¦ 99,3 ¦ 1 ¦ 86,4 ¦ 28 ¦ 82,3 ¦ 55 ¦
¦ 98,3 ¦ 2 ¦ 86,1 ¦ 29 ¦ 82,2 ¦ 56 ¦
¦ 97,4 ¦ 3 ¦ 85,9 ¦ 30 ¦ 82,1 ¦ 57 ¦
¦ 96,6 ¦ 4 ¦ 85,6 ¦ 31 ¦ 82,0 ¦ 58 ¦
¦ 96,0 ¦ 5 ¦ 85,4 ¦ 32 ¦ 81,9 ¦ 59 ¦
¦ 95,1 ¦ 6 ¦ 85,2 ¦ 33 ¦ 81,8 ¦ 60 ¦
¦ 94,3 ¦ 7 ¦ 85,0 ¦ 34 ¦ 81,7 ¦ 61 ¦
¦ 93,7 ¦ 8 ¦ 84,9 ¦ 35 ¦ 81,6 ¦ 62 ¦
¦ 93,0 ¦ 9 ¦ 84,6 ¦ 36 ¦ 81,5 ¦ 63 ¦
¦ 92,5 ¦ 10 ¦ 84,4 ¦ 37 ¦ 81,4 ¦ 64 ¦
¦ 92,0 ¦ 11 ¦ 84,3 ¦ 38 ¦ 81,3 ¦ 65 ¦
¦ 91,5 ¦ 12 ¦ 84,2 ¦ 39 ¦ 81,2 ¦ 66 ¦
¦ 91,1 ¦ 13 ¦ 84,1 ¦ 40 ¦ 81,1 ¦ 67 ¦
¦ 90,7 ¦ 14 ¦ 83,9 ¦ 41 ¦ 81,0 ¦ 68 ¦
¦ 90,5 ¦ 15 ¦ 83,8 ¦ 42 ¦ 80,9 ¦ 69 ¦
¦ 90,0 ¦ 16 ¦ 83,7 ¦ 43 ¦ 80,8 ¦ 70 ¦
¦ 89,5 ¦ 17 ¦ 83,5 ¦ 44 ¦ 80,7 ¦ 71 ¦
¦ 89,1 ¦ 18 ¦ 83,3 ¦ 45 ¦ 80,6 ¦ 72 ¦
¦ 88,8 ¦ 19 ¦ 83,2 ¦ 46 ¦ 80,5 ¦ 73 ¦
¦ 88,5 ¦ 20 ¦ 83,1 ¦ 47 ¦ 80,4 ¦ 74 ¦
¦ 88,1 ¦ 21 ¦ 83,0 ¦ 48 ¦ 80,3 ¦ 75 ¦
¦ 87,8 ¦ 22 ¦ 82,9 ¦ 49 ¦ 80,2 ¦ 76 ¦
¦ 87,5 ¦ 23 ¦ 82,8 ¦ 50 ¦ 80,1 ¦ 77 ¦
¦ 87,2 ¦ 24 ¦ 82,7 ¦ 51 ¦ 80,0 ¦ 78 ¦
¦ 87,1 ¦ 25 ¦ 82,6 ¦ 52 ¦ 79,9 ¦ 79 ¦
¦ 86,8 ¦ 26 ¦ 82,5 ¦ 53 ¦ 79,8 ¦ 80 ¦
¦ 86,6 ¦ 27 ¦ 82,4 ¦ 54 ¦ 79,7 ¦ 81 ¦
¦ 79,6 ¦ 82 ¦ 79,3 ¦ 86 ¦ 78,85 ¦ 90 ¦
¦ 79,5 ¦ 83 ¦ 79,2 ¦ 87 ¦ 78,8 ¦ 91 ¦
¦ 79,45 ¦ 84 ¦ 79,1 ¦ 88 ¦ 78,7 ¦ 92 ¦
¦ 79,4 ¦ 85 ¦ 79,0 ¦ 89 ¦ ¦ ¦
L-----------+---------+-----------+---------+-----------+----------
ОПРЕДЕЛЕНИЕ ПОКАЗАТЕЛЯ ПРЕЛОМЛЕНИЯ
(РЕФРАКТОМЕТРИЯ)
Показателем преломления (n) называют отношение скорости
распространения света в вакууме к скорости распространения света в
испытуемом веществе. Это абсолютный показатель преломления. На
практике определяют так называемый относительный показатель
преломления, т.е. отношение скорости распространения света в
воздухе к скорости распространения света в испытуемом веществе.
Показатель преломления зависит от температуры и длины волны
света, при которой проводят определение. В растворах показатель
преломления зависит также от концентрации вещества и природы
растворителя.
Рефрактометрия применяется для установления подлинности и
чистоты вещества. Метод применяют также для определения
концентрации вещества в растворе, которую находят по графику
зависимости показателя преломления от концентрации. На графике
выбирают интервал концентраций, в котором соблюдается линейная
зависимость между коэффициентом преломления и концентрацией. В
этом интервале концентрацию можно вычислить по формуле:
n - n0
X = ------,
F
где Х - концентрация раствора; n - показатель преломления
раствора; n0 - показатель преломления растворителя при той же
температуре; F - фактор, равный величине прироста показателя
преломления при увеличении концентрации на 1% (устанавливается
экспериментально).
Приборы, применяемые для определения показателя преломления,
называются рефрактометрами. Определение проводится при температуре
(20 +/- 0,3) град. C и длине волны линии D спектра натрия (589,3
нм). Показатель преломления, определенный при таких условиях,
20
обозначается индексом n .
D
Современные приборы откалиброваны таким образом, что отсчеты,
полученные по их шкалам, соответствуют показателям преломления для
D линии натрия, поэтому при проведении измерений следует соблюдать
указания в отношении соответствующего источника света, приведенные
в инструкции к приборам.
Обычно измерения показателя преломления проводят на
рефрактометрах типа Аббе, в основу которых положено явление
полного внутреннего отражения при прохождении светом границы
раздела двух сред с разными показателями преломления.
Диапазон измеряемых показателей преломления при измерении в
проходящем свете 1,3-1,7.
Точность измерения показателя преломления должна быть не ниже
-4
+/- 2 x 10 .
Могут быть использованы рефрактометры других типов с такой же
или большей точностью.
Рефрактометры юстируют по эталонным жидкостям, прилагаемым к
20
приборам, или дистиллированной воде, для которой n = 1,3330.
D
ОПРЕДЕЛЕНИЕ ОПТИЧЕСКОГО ВРАЩЕНИЯ
(ПОЛЯРИМЕТРИЯ)
Оптическое вращение - это способность вещества вращать
плоскость поляризации при прохождении через него поляризованного
света.
В зависимости от природы оптически активного вещества вращение
плоскости поляризации может иметь различное направление и
величину. Если от наблюдателя, к которому направлен свет,
проходящий через оптически активное вещество, плоскость
поляризации вращается по часовой стрелке, то вещество называют
правовращающим и перед его названием ставят знак "+", если же
плоскость поляризации вращается против часовой стрелки, то
вещество называют левовращающим и перед его названием ставят знак
"-".
Величину отклонения плоскости поляризации от начального
положения, выраженную в угловых градусах, называют углом вращения
и обозначают греческой буквой "альфа". Величина угла вращения
зависит от природы оптически активного вещества, длины пути
поляризованного света в оптически активной среде (чистом веществе
или растворе) и длины волны света. Для растворов величина угла
вращения зависит от природы растворителя и концентрации оптически
активного вещества. Величина угла вращения прямо пропорциональна
длине пути света в оптически активной среде, т.е. толщине слоя
оптически активного вещества или его раствора. Влияние температуры
в большинстве случаев незначительно.
Для сравнительной оценки способности различных веществ вращать
плоскость поляризации света вычисляют величину удельного вращения
["альфа"]. Удельное вращение - это константа оптически активного
вещества. Удельное вращение ["альфа"] определяют расчетным путем
как угол поворота плоскости поляризации монохроматического света
на пути длиной в 1 дм в среде, содержащей оптически активное
вещество, при условном приведении концентрации этого вещества к
значению, равному 1 г/мл.
Если нет специальных указаний, определение оптического
вращения проводят при температуре 20 град. С и при длине волны
линии D спектра натрия (589,3 нм). Соответствующую величину
20
удельного вращения обозначают ["альфа"] . Иногда для измерения
D
используют зеленую линию спектра ртути с длиной волны 546,1 нм.
При определении ["альфа"] в растворах оптически активного
вещества необходимо иметь в виду, что найденная величина может
зависеть от природы растворителя и концентрации оптически
активного вещества. Замена растворителя может привести к изменению
["альфа"] не только по величине, но и по знаку. Поэтому, приводя
величину удельного вращения, необходимо указывать растворитель и
выбранную для измерения концентрацию раствора.
Величину удельного вращения рассчитывают по одной из следующих
формул.
Для веществ, находящихся в растворе:
"альфа" 100
["альфа"] = -----------, (1)
lc
где "альфа" - измеренный угол вращения в градусах; l - толщина
слоя в дециметрах; с - концентрация раствора, выраженная в граммах
вещества на 100 мл раствора.
Для жидких веществ:
"альфа"
["альфа"] = -------, (2)
l"рo"
где "альфа" - измеренный угол вращения в градусах; l - толщина
слоя в дециметрах; "рo" - плотность жидкого вещества в граммах на
1 мл.
Удельное вращение определяют либо в пересчете на сухое
вещество, либо из высушенной навески, о чем в частных статьях
должно быть соответствующее указание.
Измерение величины угла вращения проводят либо для оценки
чистоты оптически активного вещества, либо для определения его
концентрации в растворе. Для оценки чистоты вещества по уравнению
(1) или (2) рассчитывают величину его удельного вращения
["альфа"]. Концентрацию оптически активного вещества в растворе
находят по формуле:
"альфа"100
c = -----------. (3)
["альфа"]l
Поскольку величина ["альфа"] постоянна только в определенном
интервале концентраций, возможность использования формулы (3)
ограничивается этим интервалом.
Измерение угла вращения проводят на поляриметре, позволяющем
определить величину угла вращения с точностью +/- 0,02 град.
Предназначенные для измерения угла вращения растворы или жидкие
вещества должны быть прозрачными. При измерении прежде всего
следует установить нулевую точку прибора или определить величину
поправки с трубкой, заполненной чистым растворителем (при работе с
растворами) или с пустой трубкой (при работе с жидкими
веществами). После установки прибора на нулевую точку или
определения величины поправки проводят основное измерение, которое
повторяют не менее 3 раз.
Для получения величины угла вращения "альфа" показания прибора,
полученные при измерениях, алгебраически суммируют с ранее
найденной величиной поправки.
ОПРЕДЕЛЕНИЯ, ОСНОВАННЫЕ НА ИЗМЕРЕНИИ
ПОГЛОЩЕНИЯ ЭЛЕКТРОМАГНИТНОГО ИЗЛУЧЕНИЯ
Фотометрические методы анализа основаны на избирательном
поглощении электромагнитного излучения анализируемым веществом и
служат для исследования строения, идентификации и количественного
анализа светопоглощающих соединений. В зависимости от используемой
аппаратуры в фотометрическом анализе различают
спектрофотометрические методы - анализ по поглощению веществами
монохроматического излучения; колориметрические и
фотоколориметрические - анализ по поглощению веществами
немонохроматического излучения.
Определения, связанные с измерением поглощения
электромагнитного излучения, основаны на двух законах. Закон
Бугера - Ламберта связывает поглощение с толщиной слоя
поглощающего вещества и выражается соотношением <*>:
-------------------------------
<*> Приведенные обозначения соответствуют ГОСТу 7601-78.
J -kb
--- = 10 ; (1)
J0
J0
lg ---- = kb, (2)
J
где J0 - интенсивность излучения, падающего на вещество; J -
интенсивность излучения, прошедшего через вещество; b - толщина
слоя вещества в сантиметрах; k - показатель поглощения <*> -
величина, обратная той толщине слоя, проходя через который поток
излучения ослабляется в 10 раз.
-------------------------------
<*> В литературе эта величина часто называется коэффициентом
погашения или коэффициентом экстинкции.
Закон Бера связывает поглощение с концентрацией поглощающего
вещества и обычно применяется для растворов:
k ="каппа"c, (3)
где с - концентрация раствора; "каппа" - показатель поглощения
раствора, концентрация которого равна единице.
На практике обычно используется объединенный закон Бугера -
Ламберта - Бера в виде:
J0
lg ---- ="каппа"cb. (4)
J
J0
Величина lg ----- носит название оптической плотности и
J
обозначается буквой D.
Величина "каппа" является специфической физической константой
для каждого вещества и может быть использована для целей
идентификации. Знание величины "каппа" позволяет определить
содержание данного вещества в растворах неизвестной концентрации
на основе измерения оптической плотности D.
Объединенный закон Бугера - Ламберта - Бера вполне справедлив
только для монохроматического излучения, поэтому строгим является
его применение в спектрофотометрии. В фотоколориметрии, где
измерения проводятся с помощью светофильтров, выделяющих
сравнительно узкий интервал длин волн, этот закон применим лишь с
большим или меньшим приближением в зависимости от степени
постоянства величины D в данном интервале длин волн.
СПЕКТРОФОТОМЕТРИЯ
Спектрофотометрия используется для идентификации соединений,
исследования состава, строения и количественного анализа
индивидуальных веществ и многокомпонентных систем. Кривая
зависимости поглощения (функция поглощения) от длины волны или
волнового числа называется спектром поглощения вещества и является
специфической характеристикой данного вещества.
В спектрофотометрических методах применяют спектрофотометры -
приборы, позволяющие проводить анализ как окрашенных, так и
бесцветных соединений по избирательному поглощению
монохроматического излучения в видимой, ультрафиолетовой и
инфракрасной областях спектра. Природа полос поглощения в
ультрафиолетовой и видимой областях спектра связана с различными
электронными переходами в поглощающих молекулах и ионах
(электронные спектры); в инфракрасной области она связана с
колебательными переходами и изменением колебательных состояний
ядер, входящих в молекулу поглощающего вещества (колебательные
спектры).
Распространенная в настоящее время аппаратура позволяет
измерять ультрафиолетовые спектры в области от 190 до 380 нм,
видимые - от 380 до 780 нм, инфракрасные спектры - от 780 до 40000
нм (40 мкм).
СПЕКТРОФОТОМЕТРИЯ В УЛЬТРАФИОЛЕТОВОЙ
И ВИДИМОЙ ОБЛАСТЯХ
Спектрофотометрические измерения в ультрафиолетовой и видимой
областях чаще всего проводят для растворов, хотя такие измерения
могут быть проведены и для веществ, находящихся в парообразном,
жидком и твердом состоянии.
Образец анализируемого вещества при спектрофотометрических
определениях обычно растворяют в соответствующем растворителе. Для
этих областей пригодны многие растворители, в том числе вода,
спирты, хлороформ, низшие углеводороды, эфиры, разведенные
растворы аммиака, едкого натра <*>, хлористоводородной или серной
кислоты. Следует использовать растворители, не содержащие
примесей, поглощающих в данной спектральной области; для
спектрофотометрии выпускаются специальные растворители,
гарантирующие отсутствие примесей.
--------------------------------
<*> Согласно принятой в стране терминологии (Химический
энциклопедический словарь, изд. Советская энциклопедия, М., 1983),
едкий натр называется гидроксид натрия, а едкое кали - гидроксид
калия.
Спектрофотометрический анализ по непосредственному измерению
оптической плотности может быть проведен для веществ, обладающих
лишь определенными особенностями строения (ароматические
соединения, соединения с сопряженными кратными связями, соединения
ряда металлов и др.).
Некоторые анализируемые вещества необходимо предварительно
перевести в соединение, поглощающее излучение.
Для определения концентрации растворов спектрофотометрическим
путем используется закон Бугера - Ламберта - Бера в форме:
1
c = --------- D. (5)
"каппа"b
В ряде случаев даже при использовании монохроматического
излучения могут наблюдаться отклонения от закона Бугера - Ламберта
- Бера, обусловленные процессами диссоциации, ассоциации и
комплексообразования. При наличии таких отклонений следует
пользоваться не формулой (5), а экспериментально найденной
зависимостью оптической плотности от концентрации.
Измерения оптической плотности D в ультрафиолетовой и видимой
области проводятся на фотоэлектрических спектрофотометрах.
Основными частями этих приборов являются: источник излучения
(лампа накаливания для видимой области, газоразрядная водородная
или дейтериевая лампа ультрафиолетовой области), монохроматор,
диспергирующая система которого основана на использовании
кварцевой призмы или дифракционной решетки, кюветное отделение, в
котором располагаются кюветы с исследуемыми веществами, приемное и
фотометрическое устройство для сравнительной оценки интенсивности
световых потоков J0 и J, основанное на использовании
фотоэлементов.
Измерительная шкала спектрофотометра проградуирована в
J
процентах пропускания Т (т.е. ---- 100) и в величинах оптической
J0 J0
плотности D (т.е. lg ----), а шкала длин волн или волновых чисел -
J
-1
в нанометрах или в см соответственно.
В процессе измерения на пути выходящего из монохроматора пучка
излучения определенной длины волны поочередно устанавливается
нулевой раствор (растворитель или раствор, содержащий те же
вещества, что и исследуемый, за исключением анализируемого
компонента), для которого Т = 100%, D = 0 и исследуемый раствор.
Для снижения величины ошибки при определении D концентрация
раствора и толщина слоя его подбираются такими, чтобы D в
исследуемой спектральной области находилось в пределах от 0,2 до
0,7. В зависимости от способности вещества к поглощению это обычно
достигается при использовании концентраций от 0,01 до 0,00001%
(кюветы с толщиной слоя 10 мм).
Показатель поглощения "каппа" вычисляют на основании измеренной
оптической плотности D для растворов с известной концентрацией по
формуле:
1
"каппа" = ----- D. (6)
cb
Концентрация "с" может быть выражена в молях на 1 л или в
граммах на 100 мл раствора. В зависимости от этого по формуле (6)
вычисляют молярный показатель поглощения или удельный показатель
поглощения.
Молярный показатель поглощения ("эпсилон") представляет собой
оптическую плотность одномолярного раствора вещества при толщине
1%
слоя 10 мм; удельный показатель поглощения (E ) - оптическую
1 см
плотность раствора, содержащего 1 г вещества в 100 мл раствора при
той же толщине слоя. Переход от удельного показателя поглощения к
молярному осуществляется по формуле:
1% M
"эпсилон" = E ----, (7)
1 см 10
где М - молекулярная масса.
1%
Если известно значение "каппа" (в форме "эпсилон" или E ),
1 см
определяютко нцентрацию исследуемых растворов по величине
оптической плотности D, пользуясь формулой (5) (при условии
подчинения закону Бера).
Для идентификации веществ в ультрафиолетовой области спектра
рекомендуется применять регистрирующие спектрофотометры.
При измерениях на разных спектрофотометрах значения характерных
длин волн могут отличаться на +/-2 нм. Если отличие превышает
указанный предел, то необходимо провести калибровку шкалы длин
волн.
При количественных определениях целесообразно использовать
такие полосы поглощения, которые отвечают следующим условиям:
1) данная полоса должна быть по возможности свободна от
наложения полос поглощения других компонентов анализируемой
системы;
2) выбранная полоса должна обладать достаточно высоким
показателем поглощения ("каппа") для индивидуального соединения.
Такие полосы называются аналитическими.
При анализе используют максимум или минимум полосы поглощения и
не следует производить измерения на участках крутого спада или
подъема кривой.
Для многокомпонентных систем выделение аналитических полос для
каждого отдельного компонента становится затруднительным, тогда
количественные определения могут быть произведены путем измерения
оптической плотности при нескольких значениях длин волн и решения
системы линейных уравнений, связывающих суммарную величину
оптической плотности смеси при данной длине волны с величиной
оптической плотности для каждого индивидуального компонента.
Например, для системы двух окрашенных веществ, спектры
поглощения которых накладываются друг на друга, определение
концентраций с1 и с2 раствора ведется при двух длинах волн по
уравнениям:
D = "эпсилон1" c1b + "эпсилон2" c2b;
"лямбда1" "лямбда1" "лямбда1"
(8)
D = "эпсилон1" c1b + "эпсилон2" c2b;
"лямбда2" "лямбда2" "лямбда2"
где D и D - измеренные экспериментально
"лямбда1" "лямбда2"
оптические плотности смеси двух веществ при длинах волн "лямбда1"
и "лямбда2"; "эпсилон1" и "эпсилон1" - молярные
"лямбда1" "лямбда2"
коэффициенты поглощения одного вещества при длинах волн "лямбда1"
и "лямбда2"; "эпсилон2" и "эпсилон2" - молярные
"лямбда1" "лямбда2"
коэффициенты поглощения второго вещества при длинах волн "лямбда1"
и "лямбда2"; b - толщина слоя вещества в сантиметрах.
Значения молярных коэффициентов поглощения определяют
экспериментально, измеряя оптические плотности стандартных
растворов каждого вещества при "лямбда1" и "лямбда2". Систему
уравнений (8) решают относительно двух неизвестных концентраций с1
и с2.
Относительная ошибка спектрофотометрических определений
индивидуальных соединений обычно не превышает 2%, при анализе
смесей ошибка определения возрастает.
В ряде случаев для идентификации и количественного определения
веществ методом спектрофотометрии требуется сравнение с
химическими стандартными образцами.
Для проверки пропускания шкалы спектрофотометров используют
стандартный образец бихромата калия. Ниже приводятся допустимые
значения оптической плотности раствора стандартного образца
бихромата калия, содержащего 60,06 мг в 1000 мл раствора серной
кислоты (0,005 моль/л), при толщине слоя 10 мм.
-------------------------------T--------T-------T-------T--------¬
¦ Длина волны ("лямбда"), нм ¦ 235 ¦ 257 ¦ 313 ¦ 350 ¦
+------------------------------+--------+-------+-------+--------+
¦ Оптическая плотность ¦ 0,748 ¦ 0,845 ¦ 0,292 ¦ 0,640 ¦
L------------------------------+--------+-------+-------+---------
СПЕКТРОФОТОМЕТРИЯ В ИНФРАКРАСНОЙ ОБЛАСТИ
Поглощением в инфракрасной области обладают молекулы, дипольные
моменты которых изменяются при возбуждении колебательных движений
ядер. Инфракрасные спектры могут быть получены в различных
агрегатных состояниях веществ и используются для идентификации,
количественного анализа, а также для исследования строения
молекул.
Измерения проводят на однолучевых и двухлучевых инфракрасных
спектрофотометрах, снабженных диспергирующими системами в виде
призм и диффракционных решеток.
Наиболее часто используется спектральная область от 2,5 до 20
-1
мкм (4000 - 500 см ).
Каждый инфракрасный спектр характеризуется серией полос
поглощения, максимумы которых определяются волновым числом
"эпсилон" или длиной волны "лямбда" и интенсивностью максимумов
поглощения.
-1
Волновое число "ни", измеряемое в обратных сантиметрах (см ),
4
10
определяется из соотношения "ни" = --------, где "лямбда" - длина
"лямбда"
волны в микрометрах (мкм).
Обычно при записи спектра на оси абсцисс откладывается в
-1
линейной шкале значение волнового числа "ни" (в см ), на оси
ординат - величина пропускания Т (в %).
Подготовку образцов к снятию инфракрасных спектров проводят по
следующим методикам.
1. Для твердых веществ. а) Пасты: тщательно смешивают 10-20 мг
твердого вещества с 1-2 каплями иммерсионной жидкости (вазелиновое
масло, полифторуглеводород, гексахлорбутадиен и др.),
приготовленную пасту сдавливают между двумя пластинками из NaCl
(или KBr) и помещают в спектрофотометр для измерения. Во второй
канал прибора помещают слой иммерсионной жидкости между
пластинками NaCl (или КВr).
б) Диски с KBr: навеску твердого вещества (1-3 мг) тщательно
смешивают в вибромельнице или в ступке со спектроскопически чистым
бромидом калия (150-200 мг) и смесь прессуют при давлении 7,5-10
т/кв. см в течение 2-5 мин. под вакуумом 2-3 мм рт. ст.
Спектр полученного образца снимают относительно воздуха или
относительно диска, приготовленного из чистого КВr, помещенного во
второй канал прибора.
2. Для жидких веществ. Тонкую пленку жидкости зажимают между
пластинками из NaCl (или КВr) или используют кюветы с малой
толщиной слоя (0,01-0,05 мм). Во второй канал прибора помещают
чистую пластинку NaCl (или КВr) удвоенной толщины или
соответствующие пустые кюветы.
3. Растворы. Раствор исследуемого образца (жидкого или
твердого) в подходящем органическом растворителе (обычно
используемые концентрации приблизительно 0,5-1,5%) вводят в кювету
с толщиной слоя 0,1-1 мм. Спектр раствора снимают относительно
чистого растворителя.
В качестве растворителей наиболее часто применяют
четыреххлористый углерод и хлороформ.
Применение инфракрасных спектров для исследования строения
веществ основано главным образом на использовании
характеристических полос поглощения (полосы, связанные с
колебаниями функциональных групп или связей в молекулах). Такими
характеристическими полосами поглощения обладают группы -ОН, -NH2,
_
-NO2, =C=O, -C=N и др.
Идентификация лекарственного вещества может быть проведена
путем сопоставления ИК-спектра исследуемого вещества с аналогичным
спектром его стандартного образца или с его стандартным спектром.
В первом случае ИК-спектры снимают последовательно на одном и том
же приборе в одинаковых условиях (агрегатное состояние образца,
концентрация вещества, скорость регистрации и т.п.).
Во втором случае следует строго руководствоваться условиями,
приведенными для стандартного спектра (концентрация вещества,
степень пропускания для основных полос и т.п.).
Обычно используют ИК-спектры, снятые с таблетками бромида калия
или с пастами (суспензиями) в вазелиновом масле.
Сопоставление ИК-спектров рекомендуется начинать с анализа
характеристических полос, которые обычно хорошо проявляются на
спектрах, и лишь при их совпадении сопоставляют низкочастотную
область.
-1
Для низкочастотного интервала 1350-400 см характерен
специфический набор полос, который называют областью "отпечатков
пальцев".
Полное совпадение полос поглощения в ИК-спектрах
свидетельствует об идентичности вещества. Полиморфные модификации
одного и того же вещества могут давать различные спектры. В этом
случае для проверки идентичности сопоставляют спектры их растворов
или, растворив каждое вещество в одном и том же растворителе,
упаривают растворитель досуха и сравнивают спектры твердых
остатков.
Наряду с положением полос поглощения существенной
характеристикой веществ является интенсивность полос поглощения,
которая может быть охарактеризована в спектрах величиной
показателя поглощения ("каппа") или величиной интегральной
интенсивности поглощения (А), равной площади огибаемой кривой
поглощения.
Интенсивности поглощения могут быть использованы для
установления строения вещества и для количественного анализа.
Колориметрия
Колориметрический метод основан на визуальном сравнении
интенсивностей окрасок растворов разных концентраций при помощи
несложных приборов: колориметрических пробирок, цилиндров с
кранами, колориметров и фотометров. В колориметрии не требуется
соблюдение закона Бера. Измерения проводят посредством следующих
операций:
а) окрашенную пробу и стандарт разбавляют в сосудах одинакового
диаметра до совпадения окрасок (метод уравнивания);
б) уравнивают окраски исследуемого окрашенного раствора с
раствором, содержащим все вещества, за исключением анализируемого,
добавляя к нему раствор этого вещества в известной концентрации
(колориметрическое титрование);
в) готовят набор стандартов с различной концентрацией вещества
и подбирают совпадение окрасок пробы и одного из стандартов (метод
стандартных серий).
Фотоколориметрия
Фотоколориметрический метод основан на измерении степени
поглощения немонохроматического света испытуемым веществом с
помощью фотоэлектроколориметров. Для определения концентраций
растворов фотоколориметрическим методом пользуются формулой (5).
Величину "каппа" и "каппа"b определяют путем проведения серии
предварительных измерений для растворов с известной концентрацией
исследуемого вещества.
_
При отсутствии линейной зависимости между "с" и D для
определения "с" следует пользоваться калибровочными графиками,
построенными для каждого определяемого вещества.
Наиболее распространенными являются две принципиальные схемы
фотоэлектроколориметров:
1) схема прямого действия с одним фотоэлементом,
предусматривающая измерение оптической плотности по силе фототока,
регистрируемой гальванометром;
2) дифференциальная схема с двумя фотоэлементами, рассчитанная
на попадание пучков света, проходящих соответственно через
испытуемый и нулевой растворы, на два разных фотоэлемента.
Фототоки уравнивают с помощью потенциометра (электрическая
компенсация) или диафрагмы, уменьшающей интенсивность одного из
световых пучков (оптическая компенсация).
По шкале потенциометра или диафрагмы отсчитывают оптическую
плотность в момент равенства фототоков, когда стрелка
регистрирующего гальванометра находится на нуле.
Относительная ошибка фотоколориметрических методов обычно не
превышает 3%, колориметрических - 5%.
Дифференциальная спектрофотометрия
и фотоколориметрия
Дифференциальный метод анализа используют для повышения
точности спектрофотометрических и фотоколориметрических измерений
при определении высоких концентраций веществ (от 10 до 100%).
Сущность метода заключается в измерении светопоглощения
анализируемого раствора относительно раствора сравнения,
содержащего определенное количество испытуемого вещества; это
приводит к изменению рабочей области шкалы прибора и снижению
относительной ошибки анализа до 0,5-1%.
Если рассматривать прохождение лучей света одинаковой
интенсивности через три кюветы, которые содержат растворитель с0 и
растворы с различной концентрацией испытуемого вещества c1 и с2,
причем с1 < с2, то интенсивность излучения прошедшего через
раствор поглощающего вещества с концентрацией с1, относительно
раствора сравнения может быть записана выражением
-kbc1
J1 = J0 x 10 , (9)
а для раствора с концентрацией с2:
-kbc2
J2 = J0 x 10 . (10)
Отношение интенсивности света, прошедшего через растворы
концентрации с2 и с1, именуемое "относительной пропускаемостью",
будет равно:
-kbc2
J2 J0 x 10 -kb(с2 - с1) -kb"ДЕЛЬТА"с
-- = ------------- = 10 = 10 ; (11)
-kbc1
J1 J0 x 10
J1
Спектроскопические
методы анализа основаны на избирательном
поглощении электромагнитного излучения
анализируемым веществом и служат для
исследования строения, идентификации
и количественного определения
светопоглощающих соединений.
В
зависимости от используемой аппаратуры
в фармацевтическом анализе различают
следующие методы анализа, основанные
на поглощении электромагнитного
излучения и испускании света:
—
спектрофотометрия в ультрафиолетовой
(УФ) и видимой областях;
—
спектрофотометрия в инфракрасной (ИК)
области;
—
атомно-эмиссионная и атомно-абсорбционная
спектроскопия (АЭС и ААС);
—
флуориметрия;
—
спектроскопия ядерного магнитного
резонанса (ЯМР).
Ряд
длин волн, для которых проводятся
измерения методами абсорбционной
спектрофотометрии, охватывает спектральную
область от коротких длин волн в УФ-области
до ИК-области. Для удобства отнесений
этот спектральный ряд делится на
следующие диапазоны длин волн: УФ (от
190 до 380 нм), видимый (от 380 до 780 нм), ИК (от
0,78 до 400 мкм).
12.1. Спектрофотометрия в ультрафиолетовой и
ВИДИМОЙ
ОБЛАСТЯХ (ОФС 42-0042-07)
Уменьшение
величины монохроматического излучения,
проходящего через гомогенную поглощающую
среду, количественно описывается законом
Бугера-Ламберта-Бера:
log10(1/T)
= A = эпсилон x c x b,
(1)
где:
T
— пропускание; T — I/I ;
o
I
— интенсивность прошедшего монохроматического
излучения;
I
— интенсивность падающего монохроматического
излучения;
o
эпсилон
— молярный показатель поглощения;
c
— молярная концентрация вещества в
растворе;
b
— длина оптического пути или толщина
слоя, в сантиметрах.
Величина
log10(1/T) носит название оптической
плотности, обозначается буквой A и
является измеряемой величиной. В
отсутствии других физико-химических
факторов измеренная оптическая плотность
(A) пропорциональна концентрации вещества
в растворе (c) и толщине слоя (b).
1%
Величина
A представляет собой удельный показатель
поглощения, т.е.
1
см
оптическую
плотность раствора вещества с
концентрацией 10 г/л (1 г/100 мл)
1%
в
кювете с толщиной слоя 1 см. Величины
A и эпсилон связаны
1
см
соотношением:
1%
10 x эпсилон
A
= ————-, (2)
1
см М.м.
где
М.м. — молекулярная масса исследуемого
вещества.
Измерение
оптической плотности. Если нет других
указаний в частной статье, измерение
оптической плотности проводят при
указанной длине волны с использованием
кювет с толщиной слоя 1 см и при температуре
(20 +/- 1) град. C по сравнению с тем же
растворителем или той же смесью
растворителей, в которой растворено
вещество. При измерении оптической
плотности раствора при данной длине
волны оптическая плотность кюветы с
растворителем, измеренная против воздуха
при той же длине волны, не должна превышать
0,4 и желательно, чтобы она была менее
0,2. Для снижения величины ошибки при
определении оптической плотности
концентрация раствора (а иногда и толщина
слоя) подбираются таким образом, чтобы
оптическая плотность в исследуемой
спектральной области находилась в
пределах от 0,2 до 0,8.
Спектр
поглощения представляют таким образом,
чтобы оптическая плотность или ее
некоторая функция были приведены по
оси ординат, а длина волны или некоторая
функция длины волны — по оси абсцисс.
Если
в частной статье для максимума поглощения
указывается только одна длина волны,
то это означает, что полученное значение
максимума не должно отличаться от
указанного более чем на +/- 2 нм.
Приборы.
Спектрофотометры, предназначенные для
измерений в ультрафиолетовой и видимой
областях спектра, состоят из оптической
системы, выделяющей монохроматическое
излучение в области от 190 до 780 нм и
обеспечивающей его прохождение через
образец, и устройства для измерения
оптической плотности.
Основными
частями этих приборов являются: источник
излучения, диспергирующий прибор (призма
или решетка), щель для выделения полосы
длин волн, кюветы для образцов, детектор
излучаемой энергии, встроенные усилители
и измерительные приборы.
Проверка
шкалы длин волн в УФ и видимой области.
Точность калибровки прибора по шкале
длин волн в спектральном ряду проверяют
по приведенным в табл. 12.1.1 спектральным
линиям водородной (Hбета) или дейтериевой
(Dбета) разрядной лампы, линиям паров
ртути (Hg) кварцево-ртутной дуговой лампы,
а также по максимумам поглощения раствора
гольмия перхлората (Ho) (готовый реактив
для калибровки спектрофотометра
представляет собой 4% раствор гольмия
оксида в 1,4 М растворе хлорной кислоты).
Допустимое отклонение составляет +/- 1
нм для ультрафиолетовой и +/- 3 нм для
видимой области.
Таблица
12.1.1
Спектральные
линии для проверки шкалы длин волн
|
241,15 |
404,66 |
|
253,7 |
435,83 |
|
287,15 |
486,0 |
|
302,25 |
486,1 |
|
313,16 |
536,3 |
|
334,15 |
546,07 |
|
361,5 |
576,96 |
|
365,48 |
579,07 |
Шкала
длин волн может быть калибрована также
при помощи подходящих стеклянных
фильтров, которые имеют фиксированные
полосы поглощения в видимой и УФ-областях,
а также стандартных стекол, содержащих
дидим (смесь празеодима и неодима), и
стекол, содержащих гольмий.
Проверка
шкалы оптической плотности. Для
проверки шкалы оптической
плотности
используют стандартные неорганические
стеклянные фильтры или
раствор
калия дихромата при длинах волн,
указанных в табл. 12.1.2, где для
каждой
длины волны приведено точное
значение удельного показателя
1%
поглощения
A и допустимые пределы.
1
см
Раствор
калия дихромата готовят следующим
образом:
от
57,0 до 63,0 мг (точная навеска) калия
дихромата, предварительно высушенного
до постоянной массы при температуре
130 град. C, растворяют в 0,005 М растворе
серной кислоты и доводят объем раствора
тем же растворителем до 1000 мл.
Таблица
12.1.2
Удельный
показатель поглощения стандартов при
различных
длинах волн
┌─────────────────────┬─────────────────────────┬─────────────────────────┐
│ Длина
волны, в │ Удельный показатель │
Допустимые пределы │
│ нанометрах
│ поглощения │ 1%
│
│ │ 1%
│ для A │
│ │ A
│ 1 см │
│ │ 1
см │ │
├─────────────────────┼─────────────────────────┼─────────────────────────┤
│ 235
│ 124,5 │ От 122,9 до 126,2 │
├─────────────────────┼─────────────────────────┼─────────────────────────┤
│ 257
│ 144,5 │ От 142,8 до 146,2 │
├─────────────────────┼─────────────────────────┼─────────────────────────┤
│ 313
│ 48,6 │ От 47,0 до 50,3 │
├─────────────────────┼─────────────────────────┼─────────────────────────┤
│ 350
│ 107,3 │ От 105,6 до 109,0 │
└─────────────────────┴─────────────────────────┴─────────────────────────┘
Предельный
уровень рассеянного света. Рассеянный
свет может быть обнаружен при данной
длине волны с использованием соответствующих
фильтров или растворов: например,
оптическая плотность раствора 12 г/л
калия хлорида в кювете с толщиной слоя
1 см при 200 нм при использовании воды в
качестве раствора сравнения должна
быть больше 2.
Разрешающая
способность (для качественного анализа).
Если есть указание в частной статье,
определяют разрешающую способность
спектрофотометра следующим образом.
Записывают спектр 0,02% (об/об) раствора
толуола в гексане. Минимально допустимое
значение отношения оптической плотности
в максимуме поглощения при 269 нм к
оптической плотности в минимуме
поглощения при 266 нм указывают в частной
статье.
Ширина
спектральной щели (для количественного
анализа). В случае использования
спектрофотометра с изменяемой шириной
спектральной щели при выбранной длине
волны возможны погрешности, связанные
с шириной этой щели. Для их исключения
ширина щели должна быть малой по сравнению
с полушириной полосы поглощения (шириной
на половине оптической плотности) и в
то же время должна быть максимально
велика для получения высокого значения
интенсивности падающего монохроматического
излучения (Io). Таким образом, ширина щели
должна быть такой, чтобы дальнейшее ее
уменьшение не изменяло величину
измеряемой оптической плотности.
Кюветы.
Допустимые отклонения в толщине слоя
используемых кювет должны быть не более
+/- 0,005 см. Кюветы, предназначенные для
испытуемого раствора и раствора
сравнения, должны иметь одинаковое
пропускание (или оптическую плотность)
при заполнении одним и тем же растворителем.
В противном случае это различие следует
учитывать.
Требования
к растворителям. Для определений,
производимых в ультрафиолетовой и
видимой областях, образец анализируемого
вещества растворяют в соответствующем
растворителе, который должен быть
оптически прозрачным в используемой
области длин волн. Для этих областей
длин волн пригодны многие растворители,
в том числе вода, спирты, хлороформ,
низшие углеводороды, эфиры и разбавленные
растворы сильных кислот и щелочей.
Идентификация
Абсорбционную
спектрофотометрию в ультрафиолетовой
и видимой областях спектра применяют
для определения подлинности лекарственных
средств путем:
—
сравнения спектров поглощения испытуемого
раствора и раствора стандартного
образца; в указанной области спектра
должно наблюдаться совпадение положений
максимумов, минимумов, плеч и точек
перегиба;
—
указания положений максимумов, минимумов,
плеч и точек перегиба; расхождение между
наблюдаемыми и указанными длинами волн
в максимумах и минимумах поглощения не
должно обычно превышать +/- 2 нм.
Возможны
и другие варианты применения, оговоренные
в частных фармакопейных статьях.
Количественное
определение
Определение
концентрации веществ спектрофотометрическим
методом основано на использовании
закона Бугера-Ламберта-Бера в форме:
A
C
= ———-, (3)
1%
A
x b
1
см
где:
C
— концентрация вещества в г/100 мл;
A
— оптическая плотность испытуемого
раствора;
1%
A
— удельный показатель поглощения
вещества;
1
см
b
— толщина поглощающего слоя, в сантиметрах.
В
ряде случаев даже при использовании
монохроматического излучения могут
наблюдаться отклонения от закона
Бугера-Ламберта-Бера, обусловленные
процессами диссоциации, ассоциации и
комплексообразования. Поэтому
предварительно следует проверить
линейность зависимости оптической
плотности раствора от концентрации в
аналитической области. При наличии
отклонений от линейной зависимости
следует пользоваться не формулой (3), а
экспериментально найденной зависимостью.
Обычно
определение концентрации
спектрофотометрическим методом проводят
с использованием стандартного образца.
Расчет концентрации основан на
использовании уравнения:
C
A
—
= —, (4)
C
A
o
o
где:
C
и C — концентрации испытуемого раствора
и раствора стандартного
o
образца соответственно;
A
и A — оптические плотности испытуемого
раствора и раствора
o
стандартного образца соответственно.
Вначале
измеряют оптическую плотность раствора
стандартного образца, приготовленного,
как указано в частной фармакопейной
статье, затем проводят измерение
оптической плотности испытуемого
раствора. Второе измерение проводят
сразу после первого, с использованием
той же кюветы, в тех же экспериментальных
условиях.
Метод
с использованием стандартного образца
является более точным и надежным.
Возможность применения значения
удельного показателя поглощения в
каждом конкретном случае следует
обосновывать. Обычно метод с использованием
значения удельного показателя поглощения
применим при допусках содержания
анализируемого вещества не менее +/- 10%
от номинального содержания.
Многокомпонентный
спектрофотометрический анализ
Многокомпонентный
спектрофотометрический анализ (анализ
смесей) применяют для одновременного
количественного определения нескольких
компонентов лекарственных средств,
каждое из которых подчиняется закону
Бугера-Ламберта-Бера.
Количественное
определение в многокомпонентном
спектрофотометрическом анализе
основывается обычно на использовании
уравнения:
m
А
= SUM E x c i = 1,…n, (5)
i
j = 1 ij j
где:
А
— оптическая плотность испытуемого
раствора при i-ой длине волны;
i
E
— показатели поглощения (зависящие
от способа выражения
Ij
концентрации)
j-го компонента образца при i-ой
аналитической длине волны;
c
— концентрация j-го компонента образца.
j
Соответствующие
методики проведения анализа и расчетные
формулы указываются в частных фармакопейных
статьях.
Производная
спектрофотометрия
В
производной спектрофотометрии исходные
спектры поглощения (нулевого порядка)
преобразуются в спектры производных
первого, второго и более высокого
порядков.
Спектр
первой производной представляет собой
график зависимости градиента кривой
поглощения (скорость изменения оптической
плотности с длиной волны, d A/d лямбда) от
длины волны.
Спектр
второй производной представляет
собой график зависимости
2
2
кривизны
спектра поглощения (d А/d лямбда ) от
длины волны. Вторая
производная
при любой длине волны связана с
концентрацией следующим
соотношением:
2
1%
2
d A
d
A 1 см
———
= ———- x c x l, (6)
2
2
d
лямбда d лямбда
где:
A
— оптическая плотность при длине волны
лямбда;
1%
A
— удельный показатель поглощения при
длине волны лямбда;
1
см
c
— концентрация вещества в растворе, в
граммах/100 мл;
l
— толщина слоя, в сантиметрах.
Производная
спектрофотометрия может быть использована
как для целей идентификации веществ,
так и их количественного определения
в многокомпонентных смесях, а также в
тех случаях, когда имеется фоновое
поглощение, вызванное присутствием
веществ, содержание которых не
регламентируется.
Приборы.
Используют спектрофотометры, отвечающие
указанным выше требованиям и оснащенные
аналоговым резистентно-емкостным
дифференцирующим модулем или цифровым
дифференциатором, или другими средствами
получения производных спектров, в
соответствии с инструкцией к прибору.
Некоторые методы получения спектров
второй производной приводят к смещению
длин волн относительно исходного
спектра, что следует учитывать там, где
это необходимо.
Разрешающая
способность. Если указано в частных
фармакопейных статьях, записывают
спектр второй производной для раствора
0,2 г/л толуола в метаноле, используя
метанол в качестве раствора сравнения.
На спектре должен присутствовать
небольшой отрицательный экстремум,
расположенный между двумя большими
отрицательными экстремумами при 261 нм
и 268 нм, в соответствии с рис. 12.1.1. (не
приводится). Если нет других указаний
в частных фармакопейных статьях,
отношение A/B должно быть не менее 0,2.
Методика.
Процедура анализа аналогична применяемой
в обычной спектрофотометрии, но вместо
оптических плотностей используют
производные. Готовят раствор испытуемого
образца, настраивают прибор в соответствии
с инструкцией производителя и рассчитывают
количество определяемого вещества, как
указано в частной фармакопейной статье.
Рис.
12.1.1. Спектр второй производной раствора
толуола
(0,2 г/л) в метаноле
Рисунок
не приводится.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Оптическая плотность раствора
Колориметрия
Из оптических методов анализа в практике аналитических лабораторий наиболее широко применяются колориметрические методы (от лат. color — цвет и греч. μετρεω — измеряю). Колориметрические методы основаны на измерении интенсивности светового потока, прошедшего через окрашенный раствор.
В колориметрическом методе используются химические реакции, сопровождающиеся изменением цвета анализируемого раствора. Измеряя светопоглощение такого окрашенного раствора или сравнивая полученную окраску с окраской раствора известной концентрации, определяют содержание окрашенного вещества в испытуемом растворе.
Существует зависимость между интенсивностью окраски раствора и содержанием в этом растворе окрашенного вещества. Эта зависимость, называемая основным законом светопоглощения (или законом Бугера—Ламберта—Бера), выражается уравнением:
I = I0 10 — ε c l
где I — интенсивность света, прошедшего через раствор; I0 — интенсивность падающего на раствор света; ε- коэффициент светопоглощения, постоянная величина для каждого окрашенного вещества, зависящая от его природы; С — молярная концентрация окрашенного вещества в растворе; l — толщина слоя светопоглощающего раствора, см.
Физический смысл этого закона можно выразить следующим образом. Растворы одного и того же окрашенного вещества при одинаковой концентрации этого вещества и толщине слоя раствора поглощают равное количество световой энергии, т. е. светопоглощение таких растворов одинаковое.
Для окрашенного раствора, заключенного в стеклянную кювету с параллельными стенками, можно сказать, что по мере увеличения концентрации и толщины слоя раствора его окраска увеличивается, а интенсивность света I, прошедшего через поглощающий раствор, уменьшается по сравнению с интенсивностью падающего света I0.
Рис.1 Прохождение света через кювету с исследуемым раствором.
Оптическая плотность раствора.
Если прологарифмировать уравнение основного закона светопоглощения и изменить знаки на обратные, то уравнение принимает вид:
Величина является очень важной характеристикой окрашенного раствора; ее называют оптической плотностью раствора и обозначают буквой A:
A = ε C l
Из этого уравнения вытекает, что оптическая плотность раствора прямо пропорциональна концентрации окрашенного вещества и толщине слоя раствора.
Другими словами, при одинаковой толщине слоя раствора данного вещества оптическая плотность этого раствора будет тем больше, чем больше в нем содержится окрашенного вещества. Или, наоборот, при одной и той же концентрации данного окрашенного вещества оптическая плотность раствора зависит только от толщины его слоя. Отсюда может быть сделан следующий вывод: если два раствора одного и того же окрашенного вещества имеют различную концентрацию, одинаковая интенсивность окраски этих растворов будет достигнута при толщинах их слоев, обратно пропорциональных концентрациям растворов. Этот вывод очень важен, так как на нем основаны некоторые методы колориметрического анализа.
Таким образом, чтобы определить концентрацию (С) окрашенного раствора, необходимо измерить его оптическую плотность (A). Чтобы измерить оптическую плотность, следует измерить интенсивность светового потока.
Интенсивность окраски растворов можно измерять различными методами. Различают субъективные (или визуальные) методы колориметрии и объективные (или фотоколориметрические).
Визуальными называются такие методы, при которых оценку интенсивности окраски испытуемого раствора делают невооруженным глазом.
При объективных методах колориметрического определения для измерения интенсивности окраски испытуемого раствора вместо непосредственного наблюдения пользуются фотоэлементами. Определение в этом случае проводят в специальных приборах — фотоколориметрах, откуда и метод получил название фотоколориметрического.
Визуальные методы
К визуальным методам относятся:
1) метод стандартных серий;
2) метод дублирования (колориметрическое титрование);
3) метод уравнивания.
Метод стандартных серий. При выполнении анализа методом стандартных серий интенсивность окраски анализируемого окрашенного раствора сравнивают с окрасками серии специально приготовленных стандартных растворов (при одинаковой толщине поглощающего слоя).
Растворы в колориметрии обычно имеют интенсивную окраску, поэтому имеется возможность определять весьма небольшие концентрации или количества веществ. Однако это может сопровождаться определенными трудностями: так навески для приготовления серии стандартных растворов могут быть очень малы. Для преодоления этих трудностей готовят стандартный раствор А достаточно высокой концентрации, например 1 мг/мл. После этого путем разбавления из раствора А готовят стандартный раствор В значительно меньшей концентрации, а из него в свою очередь готовят серию стандартных растворов.
Для этого в пробирки или кюветы одинакового размера и одинакового цвета стекла пипеткой добавляются необходимые объемы растворов реагентов в нужной последовательности. Порции растворов определяемого вещества целесообразно добавлять из бюретки, т.к. их объемы будут различны для обеспечения различных концентраций в серии стандартных растворов. При этом начальный раствор должен содержать все компоненты, кроме определяемого вещества (нулевой раствор). В исследуемый раствор добавляют растворы необходимых реагентов. Все растворы доводят до постоянного объема, а затем визуально сравнивают интенсивность окраски исследуемого раствора с растворами серии стандартных растворов. Возможно совпадение интенсивности окраски с каким-либо раствором серии. Тогда считается, сто исследуемый раствор имеет такую же концентрацию или содержит столько же определяемого вещества. Если же интенсивность окраски покажется промежуточной между соседними растворами серии, концентрация или содержание определяемого компонента считают средним арифметическим между растворами серии.
Колориметрическое титрование (метод дублирования). Этот метод основан на сравнении окраски анализируемого раствора с окраской другого раствора— контрольного. Для приготовления контрольного раствора готовят раствор, содержащий все компоненты исследуемого раствора, за исключением определяемого вещества, и все употреблявшиеся при подготовке пробы реактивы, и к нему добавляют из бюретки стандартный раствор определяемого вещества. Когда этого раствора будет добавлено столько, что интенсивности окраски контрольного и анализируемого раствора уравняются, считают, что в анализируемом растворе содержится столько же определяемого вещества, сколько его было введено в контрольный раствор.
Метод уравнивания.Этот метод основан на уравнивании окрасок анализируемого раствора и раствора с известной концентрацией определяемого вещества — стандартного раствора. Существуют два варианта выполнения колориметрического определения этим методом.
По первому варианту уравнивание окрасок двух растворов с разной концентрацией окрашенного вещества проводят путем изменения толщины слоев этих растворов при одинаковой силе проходящего через растворы светового потока. При этом, несмотря на различие концентраций анализируемого и стандартного растворов, интенсивность светового потока, проходящего через оба слоя этих растворов, будет одинакова. Соотношение между толщинами слоев и концентрациями окрашенного вещества в растворах в момент уравнивания окрасок будет выражаться уравнением:
где l1 — толщина слоя раствора с концентрацией окрашенного вещества C1, а l2-толщина слоя раствора с концентрацией окрашенного вещества C2.
В момент равенства окрасок отношение толщин слоев двух сравниваемых растворов обратно пропорционально отношению их концентраций.
На основании приведенного уравнения, измерив толщину слоев двух одинаково окрашенных растворов и зная концентрацию одного из этих растворов, легко можно рассчитать неизвестную концентрацию окрашенного вещества в другом растворе.
Для измерения толщины слоя, через который проходит световой поток, можно применять стеклянные цилиндры или пробирки, а при более точных определениях специальные приборы — колориметры.
По второму варианту, для уравнивания окрасок двух растворов с различной концентрацией окрашенного вещества, через слои растворов одинаковой толщины пропускают световые потоки различной интенсивности.
В этом случае оба раствора имеют одинаковую окраску, когда отношение логарифмов интенсивностей падающих световых потоков равно отношению концентраций.
В момент достижения одинаковой окраски двух сравниваемых растворов, при равной толщине их слоев, концентрации растворов прямо пропорциональны логарифмам интенсивностей падающего на них света.
По второму варианту определение может быть выполнено только с помощью колориметра.
Теоретические основы определения оптической плотности раствора
Любая частица, будь то молекула, атом или ион, в результате поглощения кванта света переходит на более высокий уровень энергетического состояния. Чаще всего осуществляется переход из основного в возбужденное состояние. Это вызывает появление в спектрах определенных полос поглощения.
Поглощение излучения приводит к тому, что при пропускании его через вещество интенсивность этого излучения снижается при увеличении количества частиц вещества, обладающего некоторой оптической плотностью. Этот метод исследования предложил В. М. Севергин еще в 1795 году.
Наилучшим образом этот метод годится для реакций, где определяемое вещество способно переходить в окрашенное соединение, что вызывает изменение окраски исследуемого раствора. Измерив его светопоглощение или сравнив окраску с раствором известной концентрации, несложно найти процент содержания вещества в растворе.
Основной закон светопоглощения
Суть фотометрического определения заключается в двух процессах:
- перевод определяемого вещества в поглощающее электромагнитные колебания соединение;
- замер интенсивности поглощения этих самых колебаний раствором исследуемого вещества.
Изменения в интенсивности потока света, проходящего через светопоглощающее вещество, будут вызываться также потерями света из-за отражения и рассеяния. Чтобы результат был достоверным, проводят параллельные исследования по замеру параметров при той же толщине слоя, в идентичных кюветах, с тем же растворителем. Так снижение интенсивности света зависит главным образом от концентрации раствора.
Уменьшение интенсивности света, пропущенного через раствор, характеризуют коэффициентом светопропускания (также принято называть его пропусканием) Т:
- I — интенсивность света, пропущенного через вещество;
- I0 — интенсивность падающего пучка света.
Таким образом, пропускание показывает долю непоглощенного светового потока, проходящего через изучаемый раствор. Обратный алгоритм значения пропускания называют оптической плотностью раствора (D): D = (-lgT) = (-lg) * (I / I0) = lg * (I0 / I).
Это уравнение показывает, какие параметры являются главными для исследования. К ним относится длина волны света, толщина кюветы, концентрация раствора и оптическая плотность.
Закон Бугера-Ламберта-Бера
Он является математическим выражением, отображающим зависимость уменьшения интенсивности монохроматического потока света от концентрации светопоглощающего вещества и толщины жидкостного слоя, через который он пропущен:
I = I0 * 10 -ε·С·ι , где:
- ε — коэффициент поглощения света;
- С — концентрация вещества, моль/л;
- ι —толщина слоя анализируемого раствора, см.
Преобразовав, эту формулу можно записать: I / I0 = 10 -ε·С·ι .
Суть закона сводится к следующему: различные растворы одного и того же соединения при равной концентрации и толщине слоя в кювете поглощают одинаковую часть падающего на них света.
Прологарифмировав последнее уравнение, можно получить формулу: D = ε * С * ι.
Очевидно, что оптическая плотность напрямую зависит от концентрированности раствора и толщины его слоя. Становится ясен физический смысл молярного коэффициента поглощения. Он равен D для одномолярного раствора и при толщине слоя в 1 см.
Ограничения применения закона
Этот раздел включает следующие пункты:
- Он справедлив исключительно для монохроматического света.
- Коэффициент ε связан с показателем преломления среды, особенно сильные отклонения от закона могут наблюдаться при анализе высококонцентрированных растворов.
- Температура при измерении оптической плотности должна быть постоянной (в рамках нескольких градусов).
- Световой пучок должен быть параллельным.
- рН среды должен быть постоянным.
- Закон применим для веществ, светопоглощающими центрами которых являются частицы одного вида.
Методы определения концентрации
Стоит рассмотреть метод градуировочного графика. Для его построения готовят ряд растворов (5-10) с различной концентрацией исследуемого вещества и замеряют их оптическую плотность. По полученным значениям выстраивают график зависимости D от концентрации. График является прямой линией, идущей от начала координат. Он позволяет легко определить концентрацию вещества по результатам проведенных измерений.
Также существует метод добавок. Применяется реже, чем предыдущий, но позволяет проанализировать растворы сложного состава, поскольку учитывает влияние дополнительных компонентов. Суть его состоит в определении оптической плотности среды Dx, содержащей определяемое вещество неизвестной концентрации Сх, с повторным анализом того же раствора, но с добавлением определенного количества исследуемого компонента (Сст). Величину Сх находят, используя расчеты или графики.
Условия проведения исследования
Чтобы фотометрические исследования давали достоверный результат, необходимо соблюдать несколько условий:
- реакция должна заканчиваться быстро и полностью, избирательно и воспроизводимо;
- окраска образующегося вещества должна быть устойчива во времени и не изменяться под действием света;
- исследуемое вещество берут в количестве, которого достаточно для перевода его в аналитическую форму;
- замеры оптической плотности проводят в том интервале длин волн, при котором различие в поглощении исходных реагентов и анализируемого раствора наибольшее;
- светопоглощение раствора сравнения принято считать оптическим нулем.
Способы расчета концентрации по величине аналитического сигнала
СПОСОБЫ РАСЧЕТА КОНЦЕНТРАЦИИ ПО ВЕЛИЧИНЕ АНАЛИТИЧЕСКОГО СИГНАЛА
МЕТОД ГРАДУИРОВОЧНОГО ГРАФИКА
Пример 1. При измерении оптической плотности в одинаковых условиях (длина волны 340 нм, толщина поглощающего слоя – 1,00 см) растворов калия дихромата с разной концентрацией хрома (мкг/мл) получены следующие результаты:
Изобразите примерный вид градуировочного графика; методом наименьших квадратов рассчитайте обратное уравнение градуировочного графика с = bA + а; определите концентрацию хрома (мкг/мл) в растворе Х, имеющем оптическую плотность 0,480.
Рассчитайте массу хрома (мг) в анализируемой пробе, если ее растворили в присутствии концентрированной серной кислоты в воде дистиллированной в мерной колбе объёмом 50,00 мл (раствор Х).
1. Расчет методом наименьших квадратов
2. Расчет с помощью программы Excel
длина волны 340 нм
Ответ: с = 115,72А — 8,8397 (r = 0,9941); масса хрома в пробе 2,34 мг
Пример 2. При измерении оптической плотности в одинаковых условиях (длина волны 400 нм, толщина поглощающего слоя – 1,00 см) растворов никеля (II) нитрата с разной концентрацией никеля (мг/мл) получены следующие результаты:
Изобразите примерный вид градуировочного графика; методом наименьших квадратов рассчитайте обратное уравнение градуировочного графика с = bA + а; определите концентрацию никеля (мг/мл) в растворе Х, имеющем оптическую плотность 0,350.
Рассчитайте массу никеля (мг) в анализируемой пробе, если ее количественно перенесли в мерную колбу объёмом 25,00 мл и развели водой дистиллированной в присутствии азотной кислоты до метки (раствор Х).
1. Расчет методом наименьших квадратов
2. Расчет с помощью программы Excel
Ответ: с = 42,495А — 3,8535 (r = 0,9986); масса никеля 275 мг
Пример 3. При измерении оптической плотности в одинаковых условиях (длина волны 620 нм, толщина поглощающего слоя – 1,00 см) растворов меди (II) в виде аммиачного комплекса с разной концентрацией меди (мкг/мл) получены следующие результаты:
Изобразите примерный вид градуировочного графика; методом наименьших квадратов рассчитайте обратное уравнение градуировочного графика с = bA; определите концентрацию меди (мкг/мл) в растворе Х, имеющем оптическую плотность 0,150.
Рассчитайте массу меди (мкг) в анализируемой пробе, если ее количественно перенесли в мерную колбу объёмом 25,00 мл и до метки развели водой дистиллированной в присутствии избытка аммиака (раствор Х).
1. Расчет методом наименьших квадратов
2. Расчет с помощью программы Excel
2. Расчет с помощью программы Excel
Ответ: с = 34,595А (r = 0,9985); масса меди 130 мкг.
МЕТОД ОДНОГО СТАНДАРТНОГО РАСТВОРА
Измеряют величину аналитического сигнала (yст) для раствора с известной концентрацией вещества (сст). Затем измеряют величину аналитического сигнала (yx) для раствора с неизвестной концентрацией вещества (сx). Такой способ расчёта можно использовать в том случае, если зависимость аналитического сигнала от концентрации описывается линейным уравнением без свободного члена. Концентрация вещества в стандартном растворе должна быть такой, чтобы величины аналитических сигналов, полученных при использовании стандартного раствора и раствора с неизвестной концентрацией вещества, были бы как можно ближе друг к другу.
ПРИМЕР 1. При фотометрическом определении концентрации нитрит-ионов с помощью реактива Грисса (раствора сульфаниловой кислоты и α-нафтиламина в разбавленной уксусной кислоте) было установлено, что раствор с концентрацией нитрит-ионов 2,00 мкг/мл имеет в соответствующих условиях оптическую плотность 0,300. Рассчитайте концентрацию нитрит-ионов в растворе (мкг/мл), оптическая плотность которого в таких же условиях равна 0,250. Зависимость оптической плотности от содержания аналита линейна и проходит через начало координат.
Ответ: 1,67 мкг/мл
МЕТОД ДВУХ СТАНДАРТНЫХ РАСТВОРОВ
(метод ограничивающих растворов)
Измеряют величины аналитических сигналов для стандартных растворов с двумя разными концентрациями вещества, одна из которых (с1) меньше предполагаемой неизвестной концентрации (сx), а вторая (с2) – больше. Его используют, если зависимость аналитического сигнала от концентрации описывается линейным уравнением, не проходящим через начало координат.
Пример 1. Раствор с концентрацией никеля (II) 12,00 мг/мл имеет оптическую плотность 0,350 нм, а с концентрацией 16,00 мг/мл – 0,440. Определите концентрацию никеля (мг/мл) в растворе с оптической плотностью 0,380 (все измерения проводились в одинаковых условиях: длина волны 400 нм, толщина поглощающего слоя – 1,00 см, раствор в азотной кислоте).
Ответ: 13,33 мг/мл
Используют при анализе сложных матриц, когда матричные компоненты оказывают влияний на величину аналитического сигнала и невозможно точно скопировать матричный состав образца, в случае линейной зависимости, проходящей через начало координат.
Вначале измеряют величину аналитического сигнала (yx) для пробы с неизвестной концентрацией вещества. Затем к данной пробе прибавляют некоторое точное количество определяемого вещества (стандарта) и снова измеряют величину аналитического сигнала (yдоб). Концентрацию определяемого компонента в анализируемой пробе (без учета разбавления) рассчитывают по формуле:
Для учета разбавления раствора используем формулу:
ПРИМЕР 1. Раствор с неизвестной концентрацией вещества имел оптическую плотность 0,300. К 5,00 мл такого раствора прибавили 2,00 мл раствора с концентрацией этого же вещества 40,0 мг/л. Оптическая плотность полученного раствора при измерении её в таких же условиях оказалась равна 0,500. Рассчитайте концентрацию вещества (мг/л) в исходном растворе.
1 способ: пропорционально
2 способ: преобразуем составленную пропорцию в приведенную ранее формулу
ПРИМЕР 2. Оптическая плотность раствора с неизвестным содержанием вещества равна 0,400. При добавлении к анализируемому раствору 10,0 мкг этого же вещества оптическая плотность увеличилась до 0,500. Рассчитайте массу определяемого вещества (мкг) в исходном растворе.
1 способ: пропорционально
2 способ: преобразуем составленную пропорцию в приведенную ранее формулу
источники:
http://fb.ru/article/378024/teoreticheskie-osnovyi-opredeleniya-opticheskoy-plotnosti-rastvora
http://pandia.ru/text/80/260/2737.php
Ошибки спектрофотометрических измерений определяются флуктуациями показаний на выходном приборе. Их величина зависит, в свою очередь, от стабильности источника света, флуктуаций светового пучка на пути от источника света к приемнику, шумов приемно-усилительной аппаратуры и регистрирующего прибора. Рассмотрим влияние этих источников ошибок на результаты измерений, учитывая, что при абсорбционных измерениях, в конечном итоге, существенна точность определения оптической плотности О, а не интенсивностей поглощенного и непоглощенного сигналов. Напомним, что [c.137]
Пусть чувствительность определения примесей в пробе объемом 25 мл при помощи кюветы толщиной 5 см равна 0,005 у/см . Тогда минимальное обнаруживаемое количество вещества равно 0,005 X (25/5) == 0,025 мкг, или 2,5 10 %, для пробы весом 1 г. Эта величина примерно соответствует ошибке спектрофотометрических измерений малых поглощений. [c.133]
Рассмотренные ошибки спектрофотометрического метода в основном относятся к работе прибора. Естественно, что не меньшее значение могут иметь ошибки, связанные с работой самого исследователя (точность приготовления исходных растворов, способ заполнения кювет), и с условиями протекания конкретной химиче- ской реакции (разложение реагентов, межмолекулярные взаимодействия и т. п.). Все это должно учитываться при проведении фотометрических измерений. [c.22]
Изучение факторов, влияющих на точность спектрофотометрических измерений [19] — [27], показывает, что причины ошибок в спектрофотометрии могут быть весьма разнообразны и многочисленны. Ошибки возникают, например, за счет действий оператора, условий проведения реакций, недостаточной чистоты кювет, непостоянства их установки в кюветные отделения, невоспроизводимости настройки шкалы прибора на О и 100% пропускания, непостоянства излучения источника освещения, нестабильности работы фотоэлектрической системы [24] — [27]. [c.30]
При определении Do (ВаО) на основании данных по реакций образования ВаО получаемые значения существенно зависят от принятого в расчете типа основного состояния этой молекулы. Значение, приведенное по данным работ [72, 74, 75, 396], получено для основного состояния Х 2 оно подтверждено результатами измерений методом электронного удара [73]. Расчет по результатам измерения давления пара ВаО менее надежен из-за неточности данных по АЯ (ВаО, тв.) и АЯд (Ва, тв.). В обзоре [76] принимается основное состояние и рекомендуется Do = 131 6. См. также [4, стр. 236]. Спектрофотометрические измерения [118, 119], приведшие к значению —120, содержали ошибки, см. [396]. [c.49]
На абсолютную и относительную точность (воспроизводимость) спектрофотометрических измерений влияет ряд разнообразных и часто трудно поддающихся учету факторов [23, 40]. Для количественного анализа и различных сравнительных исследований наиболее важной является воспроизводимость измерений и несущественны некоторые ошибки систематического характера, так что при разработке многих методик исследования, а также аппаратуры, исключению последних уделяется мало внимания. В связи с этим существует такое положение, что при высокой в большинстве случаев относительной точности современных спектрофотометрических измерений данные, полученные на различных приборах или в различных условиях эксперимента, часто значительно различаются. В большей части опубликованных исследований ультрафиолетовых спектров поглощения авторами не оценивается абсолютная точность измерений, а также не приводятся данные, относящиеся к аппаратуре и методике эксперимента, позволяющие провести хотя бы грубую оценку подобного рода. [c.383]
Расчет общей (максимальной) ошибки и отдельных составляющих ошибок дифференциальных спектрофотометрических измерений кобальта (14—26 г/л) в виде перхлората, измеренных по отношению к оптимальному раствору сравнения, содержащему 12 г/л Со +, приведены в табл. 7, [c.54]
Из уравнения (28) можно также сделать следующие общие заключения по точности дифференциальных спектрофотометрических измерений. Ошибка будет меньше, если [c.44]
Погрешность в определении истинной константы диссоциации слагается в основном из трех величин погрешности, вносимой спектрофотометрическими измерениями, погрешности потенциометрических измерений (если они производились) и ошибки, которая вносится принятой методикой нахождения истинной константы диссоциации по найденному значению кажущейся или концентрационной константы диссоциации. [c.93]
Ошибки при спектрофотометрических измерениях [c.137]
Первоначально рассмотрим точность анализов. Ошибка воспроизводимости, которой характеризуется точность определений атомно-абсорбционного анализа, складывается за счет двух основных операций получения поглощающего слоя и измерения поглощения спектрофотометром. Источники возникновения и величина ошибок при спектрофотометрических измерениях обсуждались ранее ( 19), причем было показано, что эти ошибки могут быть в принципе уменьшены до дробовых шумов приемника, имеющих статистическое происхождение. Случайные ошибки, связанные с получением поглощающего слоя, обусловлены следующими звеньями анализа неоднородностью образцов, дозированием проб на электрод, случайными обстоятельствами, определяющими скорость испарения элемента в кювету, колебаниями температуры кюветы и давления постороннего газа. [c.329]
Непосредственная погрешность спектрофотометрического измерения, включающая ошибки настройки прибора на О и 100% пропускания [76, 83, 88—91], погрешность отсчета по измерительному потенциометру и ошибки, связанные с нестабильностью электронной схемы [82, 92] в процессе измерения. [c.17]
Выбор условий регистрации спектров оказывает существенное влияние на результаты спектрофотометрических измерений. При выборе оптимальных условий удается свести к минимуму как систематические, так и случайные ошибки, возникающие [c.19]
Зависимость ошибки определения концентрации от величины пропускаемости света измеряемым раствором. Одна из существенных ошибок спектрофотометрических измерений возникает при отсчетах величины пропускания на крайних участках соответствующей шкалы прибора. Вследствие логарифмической формы закона поглощения наибольшая точность может быть получена при измерениях в области средних значений величины пропускания. Если предельное значение неопределенности при отсчете О величины отклонения гальванометра обозначить через (10 и предположить, что источник излучения является стабильным и что соотношение между интенсивностью падающего светового пучка и отклонением гальванометра является линейным, то [c.85]
Одновременное присутствие в растворе последовательно образующихся комплексов может привести к серьезным ошибкам при сочетании спектрофотометрических измерений с экстракцией растворителем. Только один из последовательных комплексов — электронейтральный — может экстрагироваться органическим растворителем. Различные последовательно образующиеся комплексы, которые содержат большее или меньшее количество лигандов, чем необходимо для нейтрализации положительного заряда центрального иона металла, и сами несут положительный или отрицательный заряд, останутся в водной фазе. Возможность ошибки будет наименьшей в случае комплексов, в которых для нейтрализации электрического заряда центрального иона металла и насыщения его координационной сферы требуется такое же число лигандов, как и для координации. [c.88]
Дифференциальный спектрофотометрический метод может быть применен и в тех случаях, когда имеется наложение в спектрах поглощения соединения и реагента. Тогда при измерении по отношению к одному из использованных как эталон растворов в значительной степени исключается ошибка за счет поглощения реагента. [c.480]
XI1-3-1. При каком значении Т (парциальное поглощение) в спектрофотометрических анализах будет минимальной относительная ошибка в определении концентрации (Дс/с) для данной ошибки измерения Г [c.143]
Дифференциальный метод анализа используют для повышения точности спектрофотометрических и фотоколориметрических измерений при определении высоких концентраций веществ (от 10 до 100%). Сущность метода заключается в измерении светопоглощения анализируемого раствора относительно раствора сравнения, содержащего определенное количество испытуемого вещества это приводит к изменению рабочей области шкалы прибора и снижению относительной ошибки анализа до 0,5—1%. [c.40]
Кроме того, в настоящее время разработаны спектрофотометрические методы определения большого содержания отдельных компонентов. Эти методы называют дифференциальной фотометрией. Для точного измерения в параллельном световом потоке устанавливают стандартный раствор, близкий по составу к испытуемому раствору. Таким образом, измеряется разница интенсивности двух световых потоков ошибка измерений меньше сказывается на конечном результате. Главные трудности и недостатки, по сравнению с эмиссионным спектральным анализом, связаны с затратой времени на подготовку вещества к анализу, отделение мешающих компонентов, и др. Результат зависит от выбора условий, реактивов и концентрации посторонних ионов. Групповые методы почти не разработаны, поэтому для каждого элемента необходим отдельный ход анализа. [c.9]
Другим методом определения констант является измерение возрастания растворимости в воде исследуемого вещества при различных значениях pH раствора (глава 6), Этот метод не так точен, как потенциометрический, спектрофотометрический и кондуктометрический, но бывает полезен в тех случаях (к счастью, редких), когда вещество слишком мало растворимо в воде для того, чтобы использовать потенциометрический или кондуктометрический метод, и спектр его непригоден для определения. Катализ гидролиза эфиров, дисахаридов и глюкозидов как метод измерения констант ионизации представляет лишь исторический интерес. В ряде случаев этот метод приводил к очень грубым ошибкам. [c.18]
Обычно спектрофотометрические измерения проводят в таких условиях, когда оптическая плотность исследуемого раствора лежит в 1феде-лах А = 0,2—0,8, так как именно при таких значениях оптической плотности достигается минимальная ошибка спектрофотометрических измерений. [c.528]
Общепринято, что такое ограничение не распространяется па спектрофотометрические данные, и отклонения обычно определяют как разность между необходимым и рассчитанным све-топоглощением [4, И, 12, 53, 72—74]. Обычно при определенных условиях нет необходимости использовать веса, так как в этом случае ошибки спектрофотометрического измерения преобладают над ошибками измерения концентрации [11, 12, 75]. Кроме того, показания современных спектрофотометров имеют постоянную дисперсию в некотором диапазоне значений светопоглощения (см. разд. 8.3, п. 6). Однако если измерять светопоглощение одного и того же раствора при нескольких длинах волн, то будет наблюдаться корреляция ошибок. Для математической корректности следовало бы учесть такую корреляцию, введя весовую матрицу, содержащую ковариации переменных. Тем не менее корреляцией можно пренебречь, так как спектрофотометрические ошибки начинают проявляться, когда ошибки в концентрациях составляют несколько десятых долей процента, а ошибки в измерении светопоглощения— несколько тысячных [12]. Показано [12], что даже в случае преобладания концентрационных ошибок пренебрежение корреляцией незначительно влияет на результат. [c.96]
В настоящее время метод остановленной струи широко приме-ляется для решения многих задач химической кинетики установление механизмов химической реакции, определение стадий, лимитирующих протекание реакции обнаружение промежуточных комплексов, определение кинетики ферментативных реакций, установление числа и концентрации активных центров фермента, изучение быстрых конформационны5( переходов в белках и нуклеиновых кислотах. Метод требует быстрой регистрации это единственное существенное ограничение его применимости. Особое внимание при применении метода остановленной струи необходимо уделять тер-мостатированию, так как разница в температурах в кювете наблюдения и растворе смеси реагентов может привести к большим оптическим ошибкам, затрудняющим установление механизма наблюдаемой реакции. Точность определения констант скоростей данным методом примерно такая, как и при обычных спектрофотометрических измерениях кинетики химических реакций. [c.28]
Рассмотрим в первую очередь ошибки, вытекающие из самой сущности законов поглощения излучений, и основные закономерности, установленные еще в 1937 г. Туайменом и Лотианом [19]. Найденная ими зависимость ошибки измерения А от ее абсолютного значения является определяющей в оценке ошибок спектрофотометрических измерений. [c.30]
Иногда в случае неблагоприятных условий спектрофотометрические измерения подвержены более высоким ошибкам. Например, реакцию гафния (IV) с хлораниловой кислотой в 3 М хлорной кислоте изучали спектрофотометрически в области 260—360 нм. Было измерено светопоглощение двенадцати растворов при двадцати одной длине волны [6]. На рис. 2.4 показана зависимость определяемого числа частиц от заданной ошибки матричного элемента. Результаты, полученные при исследовании хлораниловой кислоты в 3 М хлорной кислоте, также показаны на рис. 2.4. [c.41]
Определение по собственному светопоглощению. Метод основан на спектрофотометрическом измерении светопоглощения водного, раствора хлора [164, 524] или его раствора в I4 [117, 946] в УФ-области спектра (330—350 нм). Нижний предел определяемой концентрации хлора 2-10 М (1 мкг мл). Относительная ошибка при определении 10 М хлора составляет 4%, для более низких концентраций (< 10 М) ошибка увеличивается до 30— 50% [117]. [c.68]
Для индикации и регистрации показаний анализаторов все шире применяют цифровые преобразователи и цифровые регистраторы, обладающие целым рядом преимуществ перед аналоговыми, к которым в первую очередь относятся почти полное устранение ошибки считывания показаний и возможность непосредственной обработки данных на вычислительной машине. В состав цифрового регистрирующего устройства входят аналого-цифровой преобразователь, индикатор и перфоратор с логической схемой управления. Голландской фирмой Витатрон для регистрации и обработки результатов спектрофотометрических измерений выпускаются цифровой преобразователь ОНР 100 и цифровой регистратор ОКР 200. [c.135]
Спектрофотометрические измерения. На рис. 1 показаны ультрафиолетовые спектры перксеноната при различных значениях pH. Поскольку ионную силу растворов не контролировали, о результатах спектрометрических измерений можно сделать только качественные заключения. Однако очевидно, что при pH ниже И в перксенонатном растворе появляются новые формы соединений ксенона. В пределах ошибки опыта имеются две изобестических точки, что указывает на то, что во всем интервале pH имеются только две основные поглощающие формы. [c.237]
Свегла, Палл и Ердеи показали , что для вычисления ошибок спектрофотометрических измерений недостаточно учитывать только ошибки показания прибора ошибки возникают также из-за неопределенности отрезков, отсекаемых на осях координат калибровочными графиками и из-за различных наклонов последних. [c.30]
Сопоставление активационных параметров кислотного гидролиза алкилсульфатов с длинной цепью и немицеллярного этилсуль-фата показывает, что ускорение реакции при образовании мицелл связано главным образом с уменьшением энтальпии активации, а не с увеличением энтропии [212]. Этот вывод был получен с использованием потенциометрических данных. Однако энергия активации кислотного гидролиза додецилсульфата натрия, полученная из спектрофотометрических измерений, оказалась одинаковой в мицеллярных и истинных растворах (табл. 8), тогда как энтропия активации была на 6,9 энтр. ед. больше в случае мицеллярного раствора [215]. Это противоречие, вероятно, объясняется неодинаковым выбором стандарта для сравнения (раствор этилсульфата и неми-целлярный раствор додецилсульфата). Возможно также, что расхождения связаны с отклонениями температурной зависимости от уравнения Аррениуса и зависящими от температуры ошибками потенциометрического метода. [c.282]
Такой спектральный анализ требует трудно достижимой точности спектрофотометрических измерений. Он осложнен взаимным наложением аналитических полос свободных молекул и Н-комплексов, температурной зависимостью коэффициентов поглощения этих полос, неудобством и неточностью термостатирования (из-за нагрева образца излучением) и другими факторами. Ошибки спектральных определений АН составляют от 0,2 ккал1моль для прецизионных измерений до 0,5—1 ккалЫоль для обычных, но эти величины скорее характеризуют лишь невоспроизводимость измерений, а действительные неточности больше Расхождения результатов разных авторов часто намного превышает предполагаемые ошибки, достигая целых единиц ккалЫоль, или приблизительно 100% от измеряемой величины Поэтому, несмотря на большое число опубликованных данных, надежные сходящиеся значения энтальпий водородных связей известны все еще для сравнительно немногих систем . [c.139]
В работе [1049] изучены условия, при которых возможно быстрое спектрофотометрическое определение ртути в неорганических соединениях. Показано, что закон Вера выполняется для концентраций (0,5—4)-10 М Hg(II). Относительное стандартное отклонение составляет 1,8%. Изучено влияние концентрации иодида калия на определение ртути и найдено, что для 2,2-10 М Hg(II) поглощение остается неизменным, если концентрация иодида калия изменяется от 1,2 до 0,8 М. Установлено, что при pH 4 окисление Т до Тз становится заметным, однако ошибка не превышает 1%. Измерение поглощения ртутного комплекса при pH 10 дает ошибку 1%. Низкие величины оптической плотности могут быть получены при высоких pH из-за образования частиц Hg(OH) . На определение ртути данным методом оказывают влияние анионы СгО , СгзО , поглощающие в области 323 млг. Влияние СН связано с образованием частиц типа Hg( N) J4 . Ионы Ag , Сг + не влияют, если их концентрация равна 2-10 М. Но медь, платина, золото окисляют Т до и поэтому должны быть восстановлены кислым раствором НааЗгОз до анализа. Влияют на определение ртути ионы Ре(П), РЬ(П), В1(1П), Т1(1), которые дают видимые осадки в 1 М КТ при концентрации их. <1.10 М. Этот метод может быть применен в присутствии галогенидов и псевдогалогенидов. [c.105]
Одна из групп исследователей [16] вычисляла константы устойчивости, используя уравнения материального баланса. Минимизировалась сумма квадратов отклонений аналитической концентрации иона водорода. В этом случае взвешивание особенно важно, поскольку ошибка измерения pH соответствует большим отклонениям при низких значениях pH, чем при высоких [13]. Обычно взвешивание более необходимо при потенциометрических вычислениях, чем в спектрофотометрических методах 1 жно оно и тогда, когда используются отклонения функции п. Оказалось, что вычисленные веса изменяются в слишком широких пределах [26, 68, 69]. Возможно, частичной причиной этого является то, что авторы аппроксимируют данные функцией, зависимые переменные которой сами являются функциями экспериментальных наблюдений. Так, очевидно, что полная аналитическая концентрация иона водорода является экспоненциальной функцией от pH. Таким образом, условия применимости метода наименьших квадратов (разд. 4.6) выполнены не полностью, поскольку неточные зависимые переменные сопоставляются с функциями от точных значений независимых переменных. Особенно следует избегать использования отклонений функции образования п. Правильным будет применять для расчета всех потенциометрических данных функцию суммы квадратов разностей между вычисленными и наблюдаемыми э. д. с. Дополнительное преимущество такого подхода — возможность использовать единичные веса до тех пор, пока нет веских оснований полагать противное. Примером использования единичных весов служит минимизация суммы квадратов разностей меладу вычисленным и наблюдаемым объемом титрантов в процессе кислотно-основного титрования [29]. Другие исследователи также для простоты вводили допущение о единичности весовой матрицы [11, 15, 31, 51], и было сообщение, что и с весовыми коэффициентами и без них получались одни и те же значения рассчитанных констант устойчивости. [c.95]
Спектрофотометрия в ультрафиолетовой (УФ) и видимой областях (ОФС.1.2.1.1.0003.15)
Государственная фармакопея 13 издание (ГФ XIII)
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
Взамен ОФС ГФ X, ОФС ГФ XI, ОФС 42-0042-07 ГФ XII, ч.1
Спектроскопические методы анализа основаны на избирательном поглощении электромагнитного излучения анализируемым веществом и служат для исследования строения, идентификации и количественного определения светопоглощающих соединений.
В зависимости or используемой аппаратуры в фармацевтическом анализе различают следующие методы анализа, основанные на поглощении электромагнитного излучения и испускании света:
- спектрофотометрия в ультрафиолетовой (УФ) и видимой областях;
- спектрометрия в инфракрасной (ИК) области;
- атомно-эмиссионная спектрометрия (АЭС);
- атомно-абсорбционная спектроскопия (ААС);
- флуоримегрия;
- спектроскопия ядерного магнитного резонанса (ЯМР);
- масс-спектрометрия;
- рамановская спектрометрия;
- рентгеновская флуоресцентная спектрометрия;
- рентгеновская порошковая дифрактометрия.
Ряд длин волн, для которых проводятся измерения методами абсорбционной спектрофотометрии, охватывает спектральную область от коротких длин волн в УФ-области до ИК-области. Для удобства отнесений этот спектральный ряд делится на следующие диапазоны длин волн: УФ (от 190 до 380 нм), видимый (от 380 до 780 нм), ИК (от 0,78 до 400 мкм).
СПЕКТРОФОТОМЕТРИЯ В УЛЬТРАФИОЛЕТОВОЙ И ВИДИМОЙ ОБЛАСТЯХ
Уменьшение интенсивности монохроматического излучения, проходящего через гомогенную поглощающую среду, количественно описывается законом Бугера-Ламберта-Бера:
![]()
где:
Т – пропускание, отношение интенсивности светового потока, прошедшего через вещество, к интенсивности падающего на вещество светового потока: Т = I/I0;
I – интенсивность прошедшего монохроматического излучения;
I0 – интенсивность падающего монохроматического излучения;
ε – молярный показатель поглощения;
с – молярная концентрация вещества в растворе;
b – длина оптического пути или толщина слоя, в сантиметрах.
Величина log10(1/Т) носит название оптической плотности, обозначается буквой А и является измеряемой величиной. В отсутствии других физико-химических факторов измеренная оптическая плотность (А) пропорциональна концентрации вещества в растворе (с) и толщине слоя (b).
Величина ![]() представляет собой удельный показатель поглощения, т.е. оптическую плотность раствора вещества с концентрацией 10 г/л (1 г/100 мл) в кювете с толщиной слоя 1 см. Величины
представляет собой удельный показатель поглощения, т.е. оптическую плотность раствора вещества с концентрацией 10 г/л (1 г/100 мл) в кювете с толщиной слоя 1 см. Величины![]() и ε связаны соотношением:
и ε связаны соотношением:
где:
М.м. – молекулярная масса исследуемого вещества.
Измерение оптической плотности
Если нет других указаний в фармакопейной статье, измерение оптической плотности проводят при указанной длине волны с использованием кювет с толщиной слоя 1 см и при температуре (20 ± 1) °С по сравнению с тем же растворителем или той же смесью растворителей, в которой растворено вещество. При измерении оптической плотности раствора при данной длине волны оптическая плотность кюветы с растворителем, измеренная против воздуха при той же длине волны, не должна превышать 0,9 и, желательно, чтобы она была не менее 0,2.
Спектр поглощения представляют таким образом, чтобы оптическая плотность или ее некоторая функция были приведены по оси ординат, а длина волны или некоторая функция длины волны – по оси абсцисс.
Если в фармакопейной статье для максимума поглощения указывается только одна длина волны, то это означает, что полученное значение максимума не должно отличаться от указанного более чем на ± 2 нм.
Приборы
Спектрофотометры, предназначенные для измерений в ультрафиолетовой и видимой областях спектра, состоят из оптической системы, выделяющей монохроматическое излучение в области от 190 до 800 нм и обеспечивающей его прохождение через образец, и устройства для измерения оптической плотности.
Основными частями этих приборов являются: источник излучения, диспергирующий прибор (призма или решетка), щель для выделения полосы длин волн, кюветы для образцов, детектор излучаемой энергии, встроенные усилители и измерительные приборы.
Проверка шкалы длин волн в ультрафиолетовой и видимой области. Точность калибровки прибора по шкале длин волн в спектральном ряду проверяют по приведенным в табл. 1 спектральным линиям водородной (Hβ) или дейтериевой (Dβ) разрядной лампы, линиям паров ртути (Hg) кварцево-ртутной дуговой лампы, а также по максимумам поглощения раствора гольмия перхлората (Ho) (готовый реактив для калибровки спектрофотометра представляет собой 4 % раствор гольмия оксида в 14,1% растворе хлорной кислоты). Допустимое отклонение составляет ± 1 нм для ультрафиолетовой и ± 3 нм для видимой области.
Таблица 1. Максимумы поглощения для проверки шкалы длин волн
| 241,15 нм (Но) | 404,66 нм (Hg) |
| 253,7 нм (Hg) | 435,83 нм (Hg) |
| 287,15 нм (Но) | 486,0 нм (Dв) |
| 302,25 нм (Hg) | 486,1 нм (Нв) |
| 313,16 нм (Hg) | 536,3 нм (Но) |
| 334,15 нм (Hg) | 546,07 нм (Hg) |
| 361,5 нм (Но) | 576,96 нм (Hg) |
| З65,48 нм (Hg) | 579,07 нм (Hg) |
Шкала длин волн может быть калибрована также при помощи подходящих стеклянных фильтров, которые имеют фиксированные полосы поглощения в видимой и ультрафиолетовой областях, а также стандартных стекол, содержащих дидим (смесь празеодима и неодима), и стекол, содержащих гольмий.
Проверка шкалы оптической плотности. Для проверки шкалы оптической плотности используют стандартные неорганические стеклянные фильтры или раствор калия дихромата при длинах волн, указанных в табл. 2, где для каждой длины волны приведено точное значение удельного показателя поглощения ![]() и допустимые пределы.
и допустимые пределы.
Раствор калия дихромата для проверки шкалы оптической плотности при 235, 257, 313 и 350 нм готовят следующим образом: от 57,0 до 63,0 мг (точная навеска) калия дихромата, предварительно высушенного до постоянной массы при температуре 130 °С, растворяют в 0,005 М растворе серной кислоты и доводят объем раствора тем же растворителем до 1000 мл. Для проверки оптической плотности при 430 нм, растворяют 57,0-63,0 мг (точная навеска) калия дихромата в 0,005 М растворе серной кислоты и доводят объём раствора тем же растворителем до метки.
Таблица 2. Удельный показатель поглощения стандартов при различных длинах волн
Предельный уровень рассеянного света. Рассеянный свет может быть обнаружен при данной длине волны с использованием соответствующих фильтров или растворов: например, оптическая плотность раствора 12 г/л калия хлорида в кювете с толщиной слоя 1 см резко увеличивается между 220 и 200 нм и должна быть больше 2 при 198 нм при использовании воды в качестве раствора сравнения.
Разрешающая способность (для качественного анализа). Если есть указание в фармакопейной статье, определяют разрешающую способность спектрофотометра следующим образом. Записывают спектр 0,02 % (об/об) раствора толуола в гексане. Минимально допустимое значение отношения оптической плотности в максимуме поглощения при 269 нм к оптической плотности в минимуме поглощения при 266 нм указывают в фармакопейной статье.
Ширина спектральной щели (для количественного анализа). В случае использования спектрофотометра с изменяемой шириной спектральной щели при выбранной длине волны возможны погрешности, связанные с шириной этой щели. Для их исключения ширина щели должна быть малой по сравнению с полушириной полосы поглощения (шириной на половине оптической плотности) и в то же время должна быть максимально велика для получения высокого значения интенсивности падающего монохроматического излучения (I0). Таким образом, ширина щели должна быть такой, чтобы дальнейшее ее уменьшение не изменяло величину измеряемой оптической плотности.
Кюветы. Допустимые отклонения в толщине слоя используемых кювет должны быть не более ±0,005 см. Кюветы, предназначенные для испытуемого раствора и раствора сравнения, должны иметь одинаковое пропускание (или оптическую плотность) при заполнении одним и тем же растворителем. В противном случае это различие следует учитывать.
Требования к растворителям. Для определений, производимых в ультрафиолетовой и видимой областях, образец анализируемого вещества растворяют в соответствующем растворителе, который должен быть оптически прозрачным в используемой области длин волн. Для этих областей длин волн пригодны многие растворители, в том числе вода, спирты, хлороформ, низшие углеводороды, эфиры и разбавленные растворы сильных кислот и щелочей.
Идентификация
Абсорбционную спектрофотометрию в ультрафиолетовой и видимой областях спектра применяют для определения подлинности лекарственных средств путем:
- сравнения спектров поглощения испытуемого раствора и раствора стандартного образца; в указанной области спектра должно наблюдаться совпадение положений максимумов, минимумов, плеч и точек перегиба;
- указания положений максимумов, минимумов, плеч и точек перегиба спектра поглощения испытуемого раствора; расхождение между наблюдаемыми и указанными длинами волн в максимумах и минимумах поглощения не должно обычно превышать ± 2 нм.
Возможны и другие варианты применения, оговоренные в фармакопейных статьях.
Количественное определение
Определение концентрации веществ спектрофотометрическим методом основано на использовании закона Бугера-Ламберта-Бера:
В ряде случаев, даже при использовании монохроматического излучения могут наблюдаться отклонения от закона Бугера-Ламберта-Бера, обусловленные процессами диссоциации, ассоциации и комплексообразования. Поэтому предварительно следует проверить линейность зависимости оптической плотности раствора от концентрации в аналитической области. При наличии отклонений от линейной зависимости следует пользоваться не формулой (3), а экспериментально найденной зависимостью.
Обычно определение концентрации спектрофотометрическим методом проводят с использованием стандартного образца. Расчет концентрации основан на использовании уравнения:
где:
С и С0 – концентрации испытуемого раствора и раствора стандартного образца, соответственно;
А и А0 – оптические плотности испытуемого раствора и раствора стандартного образца, соответственно.
Концентрации испытуемого и стандартного раствора должны быть близки.
Вначале измеряют оптическую плотность раствора стандартного образца, приготовленного, как указано в фармакопейной статье, затем проводят измерение оптической плотности испытуемого раствора. Второе измерение проводят сразу после первого, с использованием той же кюветы, в тех же экспериментальных условиях.
Метод с использованием стандартного образца является более точным и надежным. Возможность применения значения удельного показателя поглощения в каждом конкретном случае следует обосновывать. Обычно метод с использованием значения удельного показателя поглощения применим при допусках содержания анализируемого вещества не менее ±10 % от номинального содержания.
Многокомпонентный спектрофотометрический анализ
Многокомпонентный спектрофотометрический анализ (анализ смесей) применяют для одновременного количественного определения нескольких компонентов лекарственных средств, каждое из которых подчиняется закону Бугера-Ламберта-Бера.
Количественное определение в многокомпонентном спектрофотометрическом анализе основывается обычно на использовании уравнения:
где:
Аi – оптическая плотность испытуемого раствора при i-ой длине волны;
Еij – показатели поглощения (зависящие от способа выражения концентрации) j-го компонента образца при i-ой аналитической длине волны;
cj – концентрация j-го компонента образца.
Соответствующие методики проведения анализа и расчетные формулы указываются в фармакопейных статьях.
Производная спектрофотометрия
В производной спектрофотометрии исходные спектры поглощения (нулевого порядка) преобразуются в спектры производных первого, второго и более высокого порядков.
Спектр первой производной представляет собой график зависимости градиента кривой поглощения (скорость изменения оптической плотности от длины волны, dA/dλ) от длины волны.
Спектр второй производной представляет собой график зависимости кривизны спектра поглощения (d2A/dλ2) от длины волны. Вторая производная при любой длине волны связана с концентрацией следующим соотношением:
Производная спектрофотометрия может быть использована как для целей идентификации веществ, так и для их количественного определения в многокомпонентных смесях, а также в тех случаях, когда имеется фоновое поглощение, вызванное присутствием веществ, содержание которых не регламентируется.
Приборы
Используют спектрофотометры, отвечающие указанным выше требованиям и оснащенные аналоговым резистивно-емкостным дифференцирующим модулем или цифровым дифференциатором, или другими средствами получения производных спектров, в соответствии с инструкцией к прибору. Некоторые методы получения спектров второй производной приводят к смещению длин волн относительно исходного спектра, что следует учитывать там, где это необходимо.
Разрешающая способность
Если указано в фармакопейных статьях, записывают спектр второй производной для раствора 0,2 г/л толуола в метаноле, используя метанол в качестве раствора сравнения. На спектре должен присутствовать небольшой отрицательный экстремум, расположенный между двумя большими отрицательными экстремумами при 261 нм и 268 нм, в соответствии с рис. 1. Если нет других указаний в фармакопейных статьях, отношение А/B должно быть не менее 0,2.
Методика
Процедура анализа аналогична применяемой в обычной спектрофотометрии, но вместо оптических плотностей используют производные. Готовят раствор испытуемого образца, настраивают прибор в соответствии с инструкцией производителя и рассчитывают количество определяемого вещества, как указано в фармакопейной статье.
Рисунок 1. Спектр второй производной раствора толуола (0,2 г/л) в метаноле
Ошибки спектрофотометрических измерений определяются флуктуациями показаний на выходном приборе. Их величина зависит, в свою очередь, от стабильности источника света, флуктуаций светового пучка на пути от источника света к приемнику, шумов приемно-усилительной аппаратуры и регистрирующего прибора. Рассмотрим влияние этих источников ошибок на результаты измерений, учитывая, что при абсорбционных измерениях, в конечном итоге, существенна точность определения оптической плотности О, а не интенсивностей поглощенного и непоглощенного сигналов. Напомним, что [c.137]
Пусть чувствительность определения примесей в пробе объемом 25 мл при помощи кюветы толщиной 5 см равна 0,005 у/см . Тогда минимальное обнаруживаемое количество вещества равно 0,005 X (25/5) == 0,025 мкг, или 2,5 10 %, для пробы весом 1 г. Эта величина примерно соответствует ошибке спектрофотометрических измерений малых поглощений. [c.133]
Рассмотренные ошибки спектрофотометрического метода в основном относятся к работе прибора. Естественно, что не меньшее значение могут иметь ошибки, связанные с работой самого исследователя (точность приготовления исходных растворов, способ заполнения кювет), и с условиями протекания конкретной химиче- ской реакции (разложение реагентов, межмолекулярные взаимодействия и т. п.). Все это должно учитываться при проведении фотометрических измерений. [c.22]
Изучение факторов, влияющих на точность спектрофотометрических измерений [19] — [27], показывает, что причины ошибок в спектрофотометрии могут быть весьма разнообразны и многочисленны. Ошибки возникают, например, за счет действий оператора, условий проведения реакций, недостаточной чистоты кювет, непостоянства их установки в кюветные отделения, невоспроизводимости настройки шкалы прибора на О и 100% пропускания, непостоянства излучения источника освещения, нестабильности работы фотоэлектрической системы [24] — [27]. [c.30]
При определении Do (ВаО) на основании данных по реакций образования ВаО получаемые значения существенно зависят от принятого в расчете типа основного состояния этой молекулы. Значение, приведенное по данным работ [72, 74, 75, 396], получено для основного состояния Х 2 оно подтверждено результатами измерений методом электронного удара [73]. Расчет по результатам измерения давления пара ВаО менее надежен из-за неточности данных по АЯ (ВаО, тв.) и АЯд (Ва, тв.). В обзоре [76] принимается основное состояние и рекомендуется Do = 131 6. См. также [4, стр. 236]. Спектрофотометрические измерения [118, 119], приведшие к значению —120, содержали ошибки, см. [396]. [c.49]
На абсолютную и относительную точность (воспроизводимость) спектрофотометрических измерений влияет ряд разнообразных и часто трудно поддающихся учету факторов [23, 40]. Для количественного анализа и различных сравнительных исследований наиболее важной является воспроизводимость измерений и несущественны некоторые ошибки систематического характера, так что при разработке многих методик исследования, а также аппаратуры, исключению последних уделяется мало внимания. В связи с этим существует такое положение, что при высокой в большинстве случаев относительной точности современных спектрофотометрических измерений данные, полученные на различных приборах или в различных условиях эксперимента, часто значительно различаются. В большей части опубликованных исследований ультрафиолетовых спектров поглощения авторами не оценивается абсолютная точность измерений, а также не приводятся данные, относящиеся к аппаратуре и методике эксперимента, позволяющие провести хотя бы грубую оценку подобного рода. [c.383]
Расчет общей (максимальной) ошибки и отдельных составляющих ошибок дифференциальных спектрофотометрических измерений кобальта (14—26 г/л) в виде перхлората, измеренных по отношению к оптимальному раствору сравнения, содержащему 12 г/л Со +, приведены в табл. 7, [c.54]
Из уравнения (28) можно также сделать следующие общие заключения по точности дифференциальных спектрофотометрических измерений. Ошибка будет меньше, если [c.44]
Погрешность в определении истинной константы диссоциации слагается в основном из трех величин погрешности, вносимой спектрофотометрическими измерениями, погрешности потенциометрических измерений (если они производились) и ошибки, которая вносится принятой методикой нахождения истинной константы диссоциации по найденному значению кажущейся или концентрационной константы диссоциации. [c.93]
Ошибки при спектрофотометрических измерениях [c.137]
Первоначально рассмотрим точность анализов. Ошибка воспроизводимости, которой характеризуется точность определений атомно-абсорбционного анализа, складывается за счет двух основных операций получения поглощающего слоя и измерения поглощения спектрофотометром. Источники возникновения и величина ошибок при спектрофотометрических измерениях обсуждались ранее ( 19), причем было показано, что эти ошибки могут быть в принципе уменьшены до дробовых шумов приемника, имеющих статистическое происхождение. Случайные ошибки, связанные с получением поглощающего слоя, обусловлены следующими звеньями анализа неоднородностью образцов, дозированием проб на электрод, случайными обстоятельствами, определяющими скорость испарения элемента в кювету, колебаниями температуры кюветы и давления постороннего газа. [c.329]
Непосредственная погрешность спектрофотометрического измерения, включающая ошибки настройки прибора на О и 100% пропускания [76, 83, 88—91], погрешность отсчета по измерительному потенциометру и ошибки, связанные с нестабильностью электронной схемы [82, 92] в процессе измерения. [c.17]
Выбор условий регистрации спектров оказывает существенное влияние на результаты спектрофотометрических измерений. При выборе оптимальных условий удается свести к минимуму как систематические, так и случайные ошибки, возникающие [c.19]
Зависимость ошибки определения концентрации от величины пропускаемости света измеряемым раствором. Одна из существенных ошибок спектрофотометрических измерений возникает при отсчетах величины пропускания на крайних участках соответствующей шкалы прибора. Вследствие логарифмической формы закона поглощения наибольшая точность может быть получена при измерениях в области средних значений величины пропускания. Если предельное значение неопределенности при отсчете О величины отклонения гальванометра обозначить через (10 и предположить, что источник излучения является стабильным и что соотношение между интенсивностью падающего светового пучка и отклонением гальванометра является линейным, то [c.85]
Одновременное присутствие в растворе последовательно образующихся комплексов может привести к серьезным ошибкам при сочетании спектрофотометрических измерений с экстракцией растворителем. Только один из последовательных комплексов — электронейтральный — может экстрагироваться органическим растворителем. Различные последовательно образующиеся комплексы, которые содержат большее или меньшее количество лигандов, чем необходимо для нейтрализации положительного заряда центрального иона металла, и сами несут положительный или отрицательный заряд, останутся в водной фазе. Возможность ошибки будет наименьшей в случае комплексов, в которых для нейтрализации электрического заряда центрального иона металла и насыщения его координационной сферы требуется такое же число лигандов, как и для координации. [c.88]
Дифференциальный спектрофотометрический метод может быть применен и в тех случаях, когда имеется наложение в спектрах поглощения соединения и реагента. Тогда при измерении по отношению к одному из использованных как эталон растворов в значительной степени исключается ошибка за счет поглощения реагента. [c.480]
XI1-3-1. При каком значении Т (парциальное поглощение) в спектрофотометрических анализах будет минимальной относительная ошибка в определении концентрации (Дс/с) для данной ошибки измерения Г [c.143]
Дифференциальный метод анализа используют для повышения точности спектрофотометрических и фотоколориметрических измерений при определении высоких концентраций веществ (от 10 до 100%). Сущность метода заключается в измерении светопоглощения анализируемого раствора относительно раствора сравнения, содержащего определенное количество испытуемого вещества это приводит к изменению рабочей области шкалы прибора и снижению относительной ошибки анализа до 0,5—1%. [c.40]
Кроме того, в настоящее время разработаны спектрофотометрические методы определения большого содержания отдельных компонентов. Эти методы называют дифференциальной фотометрией. Для точного измерения в параллельном световом потоке устанавливают стандартный раствор, близкий по составу к испытуемому раствору. Таким образом, измеряется разница интенсивности двух световых потоков ошибка измерений меньше сказывается на конечном результате. Главные трудности и недостатки, по сравнению с эмиссионным спектральным анализом, связаны с затратой времени на подготовку вещества к анализу, отделение мешающих компонентов, и др. Результат зависит от выбора условий, реактивов и концентрации посторонних ионов. Групповые методы почти не разработаны, поэтому для каждого элемента необходим отдельный ход анализа. [c.9]
Другим методом определения констант является измерение возрастания растворимости в воде исследуемого вещества при различных значениях pH раствора (глава 6), Этот метод не так точен, как потенциометрический, спектрофотометрический и кондуктометрический, но бывает полезен в тех случаях (к счастью, редких), когда вещество слишком мало растворимо в воде для того, чтобы использовать потенциометрический или кондуктометрический метод, и спектр его непригоден для определения. Катализ гидролиза эфиров, дисахаридов и глюкозидов как метод измерения констант ионизации представляет лишь исторический интерес. В ряде случаев этот метод приводил к очень грубым ошибкам. [c.18]
Обычно спектрофотометрические измерения проводят в таких условиях, когда оптическая плотность исследуемого раствора лежит в 1феде-лах А = 0,2—0,8, так как именно при таких значениях оптической плотности достигается минимальная ошибка спектрофотометрических измерений. [c.528]
Общепринято, что такое ограничение не распространяется па спектрофотометрические данные, и отклонения обычно определяют как разность между необходимым и рассчитанным све-топоглощением [4, И, 12, 53, 72—74]. Обычно при определенных условиях нет необходимости использовать веса, так как в этом случае ошибки спектрофотометрического измерения преобладают над ошибками измерения концентрации [11, 12, 75]. Кроме того, показания современных спектрофотометров имеют постоянную дисперсию в некотором диапазоне значений светопоглощения (см. разд. 8.3, п. 6). Однако если измерять светопоглощение одного и того же раствора при нескольких длинах волн, то будет наблюдаться корреляция ошибок. Для математической корректности следовало бы учесть такую корреляцию, введя весовую матрицу, содержащую ковариации переменных. Тем не менее корреляцией можно пренебречь, так как спектрофотометрические ошибки начинают проявляться, когда ошибки в концентрациях составляют несколько десятых долей процента, а ошибки в измерении светопоглощения— несколько тысячных [12]. Показано [12], что даже в случае преобладания концентрационных ошибок пренебрежение корреляцией незначительно влияет на результат. [c.96]
В настоящее время метод остановленной струи широко приме-ляется для решения многих задач химической кинетики установление механизмов химической реакции, определение стадий, лимитирующих протекание реакции обнаружение промежуточных комплексов, определение кинетики ферментативных реакций, установление числа и концентрации активных центров фермента, изучение быстрых конформационны5( переходов в белках и нуклеиновых кислотах. Метод требует быстрой регистрации это единственное существенное ограничение его применимости. Особое внимание при применении метода остановленной струи необходимо уделять тер-мостатированию, так как разница в температурах в кювете наблюдения и растворе смеси реагентов может привести к большим оптическим ошибкам, затрудняющим установление механизма наблюдаемой реакции. Точность определения констант скоростей данным методом примерно такая, как и при обычных спектрофотометрических измерениях кинетики химических реакций. [c.28]
Рассмотрим в первую очередь ошибки, вытекающие из самой сущности законов поглощения излучений, и основные закономерности, установленные еще в 1937 г. Туайменом и Лотианом [19]. Найденная ими зависимость ошибки измерения А от ее абсолютного значения является определяющей в оценке ошибок спектрофотометрических измерений. [c.30]
Иногда в случае неблагоприятных условий спектрофотометрические измерения подвержены более высоким ошибкам. Например, реакцию гафния (IV) с хлораниловой кислотой в 3 М хлорной кислоте изучали спектрофотометрически в области 260—360 нм. Было измерено светопоглощение двенадцати растворов при двадцати одной длине волны [6]. На рис. 2.4 показана зависимость определяемого числа частиц от заданной ошибки матричного элемента. Результаты, полученные при исследовании хлораниловой кислоты в 3 М хлорной кислоте, также показаны на рис. 2.4. [c.41]
Определение по собственному светопоглощению. Метод основан на спектрофотометрическом измерении светопоглощения водного, раствора хлора [164, 524] или его раствора в I4 [117, 946] в УФ-области спектра (330—350 нм). Нижний предел определяемой концентрации хлора 2-10 М (1 мкг мл). Относительная ошибка при определении 10 М хлора составляет 4%, для более низких концентраций (< 10 М) ошибка увеличивается до 30— 50% [117]. [c.68]
Для индикации и регистрации показаний анализаторов все шире применяют цифровые преобразователи и цифровые регистраторы, обладающие целым рядом преимуществ перед аналоговыми, к которым в первую очередь относятся почти полное устранение ошибки считывания показаний и возможность непосредственной обработки данных на вычислительной машине. В состав цифрового регистрирующего устройства входят аналого-цифровой преобразователь, индикатор и перфоратор с логической схемой управления. Голландской фирмой Витатрон для регистрации и обработки результатов спектрофотометрических измерений выпускаются цифровой преобразователь ОНР 100 и цифровой регистратор ОКР 200. [c.135]
Спектрофотометрические измерения. На рис. 1 показаны ультрафиолетовые спектры перксеноната при различных значениях pH. Поскольку ионную силу растворов не контролировали, о результатах спектрометрических измерений можно сделать только качественные заключения. Однако очевидно, что при pH ниже И в перксенонатном растворе появляются новые формы соединений ксенона. В пределах ошибки опыта имеются две изобестических точки, что указывает на то, что во всем интервале pH имеются только две основные поглощающие формы. [c.237]
Свегла, Палл и Ердеи показали , что для вычисления ошибок спектрофотометрических измерений недостаточно учитывать только ошибки показания прибора ошибки возникают также из-за неопределенности отрезков, отсекаемых на осях координат калибровочными графиками и из-за различных наклонов последних. [c.30]
Сопоставление активационных параметров кислотного гидролиза алкилсульфатов с длинной цепью и немицеллярного этилсуль-фата показывает, что ускорение реакции при образовании мицелл связано главным образом с уменьшением энтальпии активации, а не с увеличением энтропии [212]. Этот вывод был получен с использованием потенциометрических данных. Однако энергия активации кислотного гидролиза додецилсульфата натрия, полученная из спектрофотометрических измерений, оказалась одинаковой в мицеллярных и истинных растворах (табл. 8), тогда как энтропия активации была на 6,9 энтр. ед. больше в случае мицеллярного раствора [215]. Это противоречие, вероятно, объясняется неодинаковым выбором стандарта для сравнения (раствор этилсульфата и неми-целлярный раствор додецилсульфата). Возможно также, что расхождения связаны с отклонениями температурной зависимости от уравнения Аррениуса и зависящими от температуры ошибками потенциометрического метода. [c.282]
Такой спектральный анализ требует трудно достижимой точности спектрофотометрических измерений. Он осложнен взаимным наложением аналитических полос свободных молекул и Н-комплексов, температурной зависимостью коэффициентов поглощения этих полос, неудобством и неточностью термостатирования (из-за нагрева образца излучением) и другими факторами. Ошибки спектральных определений АН составляют от 0,2 ккал1моль для прецизионных измерений до 0,5—1 ккалЫоль для обычных, но эти величины скорее характеризуют лишь невоспроизводимость измерений, а действительные неточности больше Расхождения результатов разных авторов часто намного превышает предполагаемые ошибки, достигая целых единиц ккалЫоль, или приблизительно 100% от измеряемой величины Поэтому, несмотря на большое число опубликованных данных, надежные сходящиеся значения энтальпий водородных связей известны все еще для сравнительно немногих систем . [c.139]
В работе [1049] изучены условия, при которых возможно быстрое спектрофотометрическое определение ртути в неорганических соединениях. Показано, что закон Вера выполняется для концентраций (0,5—4)-10 М Hg(II). Относительное стандартное отклонение составляет 1,8%. Изучено влияние концентрации иодида калия на определение ртути и найдено, что для 2,2-10 М Hg(II) поглощение остается неизменным, если концентрация иодида калия изменяется от 1,2 до 0,8 М. Установлено, что при pH 4 окисление Т до Тз становится заметным, однако ошибка не превышает 1%. Измерение поглощения ртутного комплекса при pH 10 дает ошибку 1%. Низкие величины оптической плотности могут быть получены при высоких pH из-за образования частиц Hg(OH) . На определение ртути данным методом оказывают влияние анионы СгО , СгзО , поглощающие в области 323 млг. Влияние СН связано с образованием частиц типа Hg( N) J4 . Ионы Ag , Сг + не влияют, если их концентрация равна 2-10 М. Но медь, платина, золото окисляют Т до и поэтому должны быть восстановлены кислым раствором НааЗгОз до анализа. Влияют на определение ртути ионы Ре(П), РЬ(П), В1(1П), Т1(1), которые дают видимые осадки в 1 М КТ при концентрации их. <1.10 М. Этот метод может быть применен в присутствии галогенидов и псевдогалогенидов. [c.105]
Одна из групп исследователей [16] вычисляла константы устойчивости, используя уравнения материального баланса. Минимизировалась сумма квадратов отклонений аналитической концентрации иона водорода. В этом случае взвешивание особенно важно, поскольку ошибка измерения pH соответствует большим отклонениям при низких значениях pH, чем при высоких [13]. Обычно взвешивание более необходимо при потенциометрических вычислениях, чем в спектрофотометрических методах 1 жно оно и тогда, когда используются отклонения функции п. Оказалось, что вычисленные веса изменяются в слишком широких пределах [26, 68, 69]. Возможно, частичной причиной этого является то, что авторы аппроксимируют данные функцией, зависимые переменные которой сами являются функциями экспериментальных наблюдений. Так, очевидно, что полная аналитическая концентрация иона водорода является экспоненциальной функцией от pH. Таким образом, условия применимости метода наименьших квадратов (разд. 4.6) выполнены не полностью, поскольку неточные зависимые переменные сопоставляются с функциями от точных значений независимых переменных. Особенно следует избегать использования отклонений функции образования п. Правильным будет применять для расчета всех потенциометрических данных функцию суммы квадратов разностей между вычисленными и наблюдаемыми э. д. с. Дополнительное преимущество такого подхода — возможность использовать единичные веса до тех пор, пока нет веских оснований полагать противное. Примером использования единичных весов служит минимизация суммы квадратов разностей меладу вычисленным и наблюдаемым объемом титрантов в процессе кислотно-основного титрования [29]. Другие исследователи также для простоты вводили допущение о единичности весовой матрицы [11, 15, 31, 51], и было сообщение, что и с весовыми коэффициентами и без них получались одни и те же значения рассчитанных констант устойчивости. [c.95]
МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РОССИЙСКОЙ ФЕДЕРАЦИИ
УРАЛЬСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
МЕТОДИЧЕСКИЕ УКАЗАНИЯ К ЛАБОРАТОРНЫМ РАБОТАМ «СПЕКТРОФОТОМЕТРИЧЕСКИЙ АНАЛИЗ» ПО СПЕЦКУРСУ «ОПТИЧЕСКИЕ МЕТОДЫ АНАЛИЗА» ДЛЯ СТУДЕНТОВ 4 КУРСА ХИМИЧЕСКОГО ФАКУЛЬТЕТА. СПЕЦИАЛЬНОСТЬ «ХИМИЯ»
ЕКАТЕРИНБУРГ 2005
Методические указания подготовлены кафедрой аналитической химии
Составитель:
Буянова Е.С.
Емельянова Ю.В.
Уральский государственный университет
2005
ОТ СОСТАВИТЕЛЯ
При изучении специального курса «Оптические методы анализа» студенты осваивают наиболее распространенные методы качественного и количественного эмиссионного спектрального анализа, спектрофотометрического анализа. В настоящих методических указаниях описаны лабораторные работы, выполнение которых позволяет студентам получить определенные навыки в проведении точного аналитического эксперимента и обработке экспериментальных данных. В руководстве описана необходимая аппаратура, используемая при различных измерениях, правила ее эксплуатации и порядок измерений. Описанию лабораторных методик предшествует краткое изложение теоретических основ метода, облегчающее выполнение конкретных аналитических задач. Методические указания разделены на две части: спектральный анализ и спектрофотометрический анализ.
Выполняя практические работы, студент должен выполнять следующие правила:
- Ознакомиться с инструкцией по технике безопасности при работе в лаборатории
- Ознакомиться с описанием конкретной работы, уяснить цель работы и методику ее выполнения.
- Ознакомиться с описанием прибора, на котором выполняется работа и методикой измерений на нем.
- Приготовить в строгом соответствии с методикой необходимые приборы, материалы, реактивы и посуду.
- Получить у преподавателя или дежурного лаборанта разрешение на включение прибора.
- Получить у преподавателя или дежурного лаборанта контрольную задачу и необходимые материалы.
- По окончании работы выключить прибор, привести в порядок и сдать рабочее место лаборанту.
- Оформить и сдать преподавателю отчет о проделанной работе.
Краткая теория
Методы анализа, основанные на измерении поглощения излучения молекулярной средой в видимой и УФ-областях, называют спектрофотометрическими. Единой теоретической базой всех разновидностей спектрофотометрии является закон Бугера – Ламберта – Бера:
A = k ∙ l ∙ c
Коэффициент поглощения k в данном выражении равен оптической плотности при единичной концентрации и толщине слоя и в зависимости от способа выражения последних, может иметь разные единицы измерения. В количественном анализе обычно выражают концентрацию в молях на литр, а толщину слоя – в сантиметрах, тогда k называют молярным коэффициентом поглощения и обозначают буквой . Молярный коэффициент поглощения – важнейшая молекулярная характеристика, не зависящая от концентрации и толщины поглощающего слоя. Она может служить объективным критерием чувствительности фотометрического определения. Светопоглощение подчиняется также закону аддитивности: оптическая плотность смеси веществ равна сумме оптических плотностей каждого из них (при условии подчинения закону Бугера – Ламберта – Бера). Для одной и той же длины волны и толщины слоя для смеси веществ
A = ε1 ∙l1 ∙c1 + ε2 ∙l2 ∙c2 + …εn ∙ln ∙cn
Отклонения от закона Бугера – Ламберта – Бера. Поведение поглощающих свет систем подчиняется закону Бугера – Ламберта – Бера при определенных условиях. При нарушении этих условий молярный коэффициент поглощения изменяется. Если он уменьшается, наблюдаются отрицательные отклонения от закона, если возрастает – положительные отклонения. Причины отклонений от основного закона светопоглощения могут быть кажущимися и истинными. Кажущиеся причины, обусловленные немонохроматичностью светового потока, рассеянием света и случайными излучениями, называют инструментальными, а вызванные химическими взаимодействиями – химическими. Истинные причины связаны с изменениями в окружении поглощающих частиц при повышении концентрации и с допущениями, сделанными при выводе основного закона светопоглощения.
Представление спектров поглощения. Спектр поглощения вещества – графическое изображение распределения поглощаемой энергии по длинам волн. Способы представления спектров различаются величинами, откладываемыми по осям абсцисс и ординат. По оси ординат откладывают оптическую плотность, логарифм оптической плотности, пропускание (в долях пропускания или в процентах). По оси абсцисс откладывают длину волны, частоту, волновое число. Выбор той или иной величины определяется стоящими перед исследователем задачами, областью спектра; величиной поглощения и т. п.
Для целей качественного анализа удобно представить спектр в координатах длина волны – молярный коэффициент поглощения. В случае подчинения закону Бугера – Ламберта – Бера независимо от концентрации спектр сохраняет свой вид. При отклонениях от закона наблюдается смещение максимума поглощения или другие изменения.
Для выявления всех характерных особенностей спектральных кривых их можно продифференцировать (производная спектрофотометрия). Тогда спектр будет представлять собой зависимость первой, второй и т.д. производных оптической плотности от (): ![]() Для первой и вообще нечетных производных вместо обычной полосы поглощения получаются кривые вида дисперсионной функции. Эти производные позволяют легче выявить и определить положение точек перегиба и замаскированных пиков, поскольку в максимумах поглощения
Для первой и вообще нечетных производных вместо обычной полосы поглощения получаются кривые вида дисперсионной функции. Эти производные позволяют легче выявить и определить положение точек перегиба и замаскированных пиков, поскольку в максимумах поглощения ![]() . Вторая и последующие четные производные дают пики, совпадающие по положению с максимумом полосы поглощения по первой производной. Эти пики резче, чем исходная полоса, за счет чего может быть получено более высокое разрешение. Дифференциальный спектр можно получить также с помощью двухволнового спектрофотометра, в котором через одну и ту же кювету одновременно проходят два потока излучения с разными длинами волн.
. Вторая и последующие четные производные дают пики, совпадающие по положению с максимумом полосы поглощения по первой производной. Эти пики резче, чем исходная полоса, за счет чего может быть получено более высокое разрешение. Дифференциальный спектр можно получить также с помощью двухволнового спектрофотометра, в котором через одну и ту же кювету одновременно проходят два потока излучения с разными длинами волн.
Измерение поглощения. Прибор для измерения светопоглощения состоит из ряда узлов, соединенных в определенной последовательности. Прибор должен выполнять две основные задачи: 1) разложить полихроматический свет по длинам волн и выделить нужный интервал длин волн; 2) оценить поглощение света веществом при выбранной длине волны.
Каждый прибор включает: источник излучения, устройство для выделения нужного интервала длин волн (монохроматор или светофильтр), кюветное отделение, детектор, преобразователь сигнала, индикатор сигнала (шкалу или цифровой счетчик). Порядок расположения узлов может быть разным (например, монохроматор может стоять до кюветы или после нее).
Типичные источники излучения в спектрофотометрии – лампа накаливания с вольфрамовой нитью, дейтериевая (водородная) лампа или галогенокварцевая лампа. Эти источники излучают в широкой области спектра, поэтому излучение нужно монохроматизировать. Приборы, в которых для монохроматизации используют монохроматоры, называют спектрофотометрами (отсюда – спектрофотометрический метод анализа), а те, в которых необходимый интервал длин волн выделяют светофильтром, называют фотоэлектроколориметрами (ФЭК).
В абсорбционной спектроскопии измеряется не абсолютное значение оптической плотности, а разность оптических, плотностей исследуемого раствора и раствора, оптическая плотность которого принята за нуль (раствор сравнения). Кювета, в которую помещают исследуемый раствор, называется рабочей, а кювета для раствора сравнения – кюветой сравнения. Обе кюветы должны быть по возможности идентичны. Основное требование к кюветам – прозрачность в области спектра, в которой ведется измерение оптической плотности. Для работы в видимой области кюветы изготовляют из стекла. В ультрафиолетовой области стекло непригодно: кюветы делают из кварца. По форме кюветы бывают прямоугольными и цилиндрическими.
Для некоторых работ требуются кюветы специальной конструкции. Для исследования кинетики реакций применяют термостатированные кюветы (с «рубашкой» из стекла, через которую циркулирует вода с определенной температурой). В автоматических установках используют проточные кюветы.
Для приема сигнала в видимой и УФ-областях обычно применяют фотоэлементы и фотоумножители. Наиболее употребительны сурьмяно – цезиевые (в диапазоне 180 – 650 нм) и кислородно – цезиевые (в диапазоне 600 – 1100 нм) фотоэлементы.
В зависимости от способа измерения различают одно- и двухлучевые приборы, от способа монохроматизации – фотоэлектроколориметры и спектрофотометры, от способа регистрации – визуальные, регистрирующие и нерегистрирующие приборы.
Фотоэлектроколориметры имеют простую конструкцию и пригодны для измерений в видимой и ближней (до 300 нм) УФ-областях. Оптические детали этих приборов изготовлены из стекла или просветленного стекла. Фотоэлектроколориметры используют чаще всего для проведения серийных определений концентраций веществ.
Спектрофотометры имеют более сложную конструкцию и часто снабжены электронными устройствами (усилителями фототока, дисплеями). Оптические детали изготовлены из кварца. Спектрофотометры применяют для получения спектров поглощения, а также для измерений концентраций веществ с узкой полосой поглощения или веществ с близкими длинами волн поглощения.
Аппаратура и принадлежности для фотометрического анализа
КОЛОРИМЕТР ФОТОЭЛЕКТРИЧЕСКИЙ
КОНЦЕНТРАЦИОННЫЙ КФК-2
1. Описание прибора
Принцип измерения коэффициента пропускания состоит в том, что на фотоприемник направляются поочередно различные световые потоки: полный и прошедший через исследуемую среду, и определяется отношение этих потоков.
Принципиальная оптическая схема фотометра приведена на рис. 1.

Рис. 1. Оптическая схема фотоэлектроколориметра КФК-2
2. Подготовка к работе
2. 1. Колориметр включить в сеть за 15 минут до начала измерений. Во время прогрева кюветное отделение должно быть открыто (при этом шторка перед фотоприемниками перекрывает световой пучок).
2. 2. Ввести необходимый по роду измерения цветной светофильтр.
2.3. Установить минимальную чувствительность колориметра. Для этого ручку «ЧУВСТВИТЕЛЬНОСТЬ» (рис. 2) необходимо установить в положение 1, ручку «УСТАНОВКА 100 ГРУБО» – в крайнее левое положение.
2. 4. Перед измерениями и при переключении фотоприемников проверить установку стрелки колориметра на «0» по шкале коэффициентов пропускания Т при открытом кюветном отделении. При смещении стрелки от нулевого положения, ее подводят к нулю с помощью потенциометра «НУЛЬ», выведенного под шлиц.

Рис. 2. Внешний вид колориметра КФК-2
3. Порядок работы
3. 1. Измерение коэффициента пропускания
3. 1. 1. В световой пучок поместить кювету с растворителем или контрольным раствором, по отношению к которому производятся измерения.
3. 1. 2. Закрыть крышку кюветного отделения.
3. 1. 3. Ручками «ЧУВСТВИТЕЛЬНОСТЬ» и «УСТАНОВКА 100 ГРУБО» и «ТОЧНО» установить отсчет «100» по шкале колориметра.
3. 1. 4. Затем, поворотом ручки кювету с растворителем или контрольным раствором заменить кюветой с исследуемым раствором.
3. 1. 5. Снять отсчет по шкале колориметра, соответствующей коэффициенту пропускания исследуемого раствора в процентах. Для регистрирующего прибора типа M 907-10 отсчет снимают по шкале коэффициентов пропускания Т в процентах, или по шкале Д – в единицах оптической плотности. Абсолютная погрешность измерения коэффициента пропускания не превышает 1%.
3. 1. 6. Измерение провести 3 – 5 раз и окончательное значение измеренной величины определить как среднее арифметическое из полученных значений.
3. 2. Определение концентрации вещества в растворе
При определении концентрации вещества в растворе следует соблюдать следующую последовательность в работе:
– выбор светофильтра;
– выбор кюветы;
– построение градуировочной зависимости для данного вещества;
– измерение оптической плотности исследуемого раствора и определение концентрации вещества в растворе.
3. 2. 1. Выбор светофильтра.
Наличие в колориметре узла светофильтров и набора кювет позволяет подобрать такое их сочетание, при котором погрешность в определения концентрации будет наименьшей.
Выбор светофильтра проводят следующим образом. Наливают раствор в кювету (о выборе размера кювет см. ниже) и определяют оптическую плотность для всех светофильтров.
По полученным данным строят зависимость, откладывая по горизонтальной оси длины волн, соответствующие максимуму коэффициента пропускания светофильтров, указанные в описании колориметра, а по вертикальной оси – соответствующие значения оптической плотности раствора. Отмечают тот участок кривой, для которого выполняются следующие условия:
– оптическая плотность имеет максимальную величину;
– ход кривой примерно параллелен горизонтальной оси,
т. е. оптическая плотность мало зависит от длины волн. Светофильтр для работы выбирают так, чтобы длина волны, соответствующая максимуму коэффициента пропускания светофильтра, приходилась на отмеченный выше участок спектральной кривой испытуемого раствора.
Если эти условия выполняются для нескольких светофильтров, то выбирают тот из них, для которого чувствительность колориметра выше.
3. 2. 2. Выбор кюветы.
Относительная ошибка определения концентрации раствора будет различной при работе на разных участках шкалы колориметра и достигает минимума при значении оптической плотности 0.4. Поэтому при работе на колориметре рекомендуется, путем соответствующего выбора кювет, работать вблизи указанного значения оптической плотности.
Предварительный выбор кювет проводится визуально, соответственно интенсивности окраски раствора. Если раствор интенсивно окрашен (темный), следует пользоваться кюветами с малой рабочей длиной. В случае слабо окрашенных растворов рекомендуется работать с кюветами с большой рабочей длиной.
В предварительно подобранную кювету наливают раствор и измеряют его оптическую плотность, вводя в ход лучей соответствующий для данного раствора светофильтр.
Второе условие может для некоторых растворов не иметь места, тогда при выборе светофильтра ограничиваются выполнением первого условия.
При измерении ряда растворов кювету заполняют раствором средней концентрации. Если полученное значение оптической плотности составляет примерно 0.3 – 0.5 – выбирают данную кювету для работы с этим раствором. В том случае, когда это условие не выполняется, следует испробовать другую кювету. Если величина измеренной оптической плотности больше 0.5 – 0.6, берут кювету меньшей рабочей длины, если величина оптической плотности меньше 0.3 – 0.2, следует выбрать кювету с большей рабочей длиной.
3. 2. 3. Построение градуировочного графика для данного вещества.
Построение градуировочного графика проводят следующим образом. Готовят ряд растворов данного вещества с известными концентрациями, охватывающими область возможных изменений концентраций этого вещества в исследуемом растворе.
Измеряют оптические плотности всех растворов и строят градуировочный график, откладывая по горизонтальной оси известные концентрации, а по вертикальной соответствующие им значения оптической плотности.
3. 2. 4. Определение концентрации вещества в растворе.
По градуировочному графику в дальнейшем определяют неизвестную концентрацию вещества в исследуемых растворах. Для этого раствор наливают в ту же кювету, для которой построена градуировочная зависимость, и, выбрав тот же светофильтр, определяют оптическую плотность раствора. Затем по градуировочному графику находят концентрацию, соответствующую измеренному значению оптической плотности.
Примечания: 1. Часто в работе бывает удобнее пользоваться градуировочными таблицами, которые составляются по данным градуировочной зависимости.
2. Градуировочный график следует время от времени проверять.
Фотометр фотоэлектрический КФК-3
Содержание
- § 126. Точность измерений в спектрофотометрическом методе
- «ТЕСТОВЫЕ ЗАДАНИЯ К ЗАЧЕТУ ДЛЯ СТУДЕНТОВ 3 КУРСА ОЧНОГО ОТДЕЛЕНИЯ ПО ДИСЦИПЛИНЕ «МЕТОДЫ ФАРМАЦЕВТИЧЕСКОГО АНАЛИЗА» 1. По принципу взаимодействия разделяемых компонентов . »
- ТЕСТОВЫЕ ЗАДАНИЯ К ЗАЧЕТУ ДЛЯ СТУДЕНТОВ 3 КУРСА ОЧНОГО ОТДЕЛЕНИЯ
- ПО ДИСЦИПЛИНЕ «МЕТОДЫ ФАРМАЦЕВТИЧЕСКОГО АНАЛИЗА»
§ 126. Точность измерений в спектрофотометрическом методе
В фотометрическом анализе может быть две группы ошибок:
1) ошибки, связанные с получением химического соединения, поглощающего излучения определенной длины волны;
2) ошибки, связанные с процессом измерения оптической плотности раствора.
Источники первой группы ошибок многочисленны. В первую очередь эти ошибки зависят от правильности выбора оптимальных условий и четкости проведения фотометрической реакции.
Ошибка измерения оптической плотности существенно зависит от значения D.
Из формулы D = IgZo/// следует, что оптическая плотность может принимать значения от 0 (U = h) до со (Z1 = O). Однако из графической зависимости D от h (рис. 71) видно, что область оптических плотностей, имеющих практическое значение,
ограничена участком, в пределах которого изменение U вызывает примерно одинаковое изменение D.
Относительная ошибка фотометрического измерения Д описывается уравнением:
где AD — наименьшая ошибка в измерении D, являющаяся функцией самой измеряемой величины; ЛС — ошибка определения концентрации; AT — наименьшее доступное измерению на данном приборе изменение пропускания (T).


Рис. 71. Зависимость оптической плотности D от интенсивности потока излучения, прошедшего через раствор 1[.
Рис. 72. Зависимость относительной ошибки фотометрического измерения Д от величины оптической плотности:
/ — кривая Шмидта (рассчитана теоретически); 2 —кривая Комаря и Самойлова получена экспериментально по данным измерения D на спектрофотометре (СФ-4).
Эта зависимость изображена графически на рис. 72, откуда видно, что Д имеет минимум при Z) = 0,43 (T = 36,8%) и возрастает незначительно в интервале, равном 2ДМИН, что соответствует примерно интервалу D от 0,12 до 1,2 или Г —от 76 до 16%. Однако данная зависимость была получена при двух основных допущениях: закон Бугера — Ламберта — Бера соблюдается при любых значениях D (от 0 до со), т. е. при любых концентрациях, и ошибка измерения пропускания AT не зависит от величины Т. Оба эти предположения не могут быть справедливыми для всех значений D, так как закон часто не соблюдается при больших значениях D и ошибка в отсчете T неодинакова при различных значениях этой величины.
В работах Комаря с сотр. на основании экспериментальной проверки закономерности было показано, что интервал D может быть расширен главным образом в сторону больших значений D вплоть до D
2,0 (см. рис. 72, кривая 2) без значительного снижения точности определения.
Оптимальные пределы D, рассмотренные выше, относятся к абсолютному методу фотометрического анализа, когда D испытуемого раствора измеряется по отношению к растворителю. Чтобы иметь возможность использовать спектрофотометрические .методы для определения с достаточной точностью больших количеств веществ, необходимо измерять значения fl>2 с достаточно боль-
шой точностью. В таких случаях можно применить так называемый дифференциальный (сравнительный) метод фотометрии, где в качестве нулевого раствора используется один из эталонных растворов. Величина измеренной D0th представляет собой разность между абсолютными значениями оптических плотностей исследуемого раствора (/Зиссл) и эталоннного раствора (D0). Тогда уравнение относительной ошибки измерения D можно записать следующим образом:
Источник
«ТЕСТОВЫЕ ЗАДАНИЯ К ЗАЧЕТУ ДЛЯ СТУДЕНТОВ 3 КУРСА ОЧНОГО ОТДЕЛЕНИЯ ПО ДИСЦИПЛИНЕ «МЕТОДЫ ФАРМАЦЕВТИЧЕСКОГО АНАЛИЗА» 1. По принципу взаимодействия разделяемых компонентов . »
ТЕСТОВЫЕ ЗАДАНИЯ К ЗАЧЕТУ ДЛЯ СТУДЕНТОВ 3 КУРСА ОЧНОГО ОТДЕЛЕНИЯ
ПО ДИСЦИПЛИНЕ «МЕТОДЫ ФАРМАЦЕВТИЧЕСКОГО АНАЛИЗА»
1. По принципу взаимодействия разделяемых компонентов смеси со структурными
компонентами неподвижной фазы выделяют хроматографию:
2. По расположению неподвижной фазы выделают хроматографию:
а. Колоночную б. Бумажную в. Препаративную г. Аналитическую д. Плоскостную Правильные ответы: а, д.
3. По сфере применения выделают хроматографию:
а. Осадочную б. Препаративную в. Тонкослойную г. Распределительную д. Аналитическую е. Разделительную Правильные ответы: б, д.
4. Сопоставьте вид хроматографии и принцип взаимодействия разделяемых компонентов и неподвижной фазы, на котором он основан:
5. Лигандообменная а. Образование малорастворимых соединений с различной степенью растворимости б. Взаимодействие «антиген-антитело» в. Образование комплексных соединений с различной константой нестойкости г. Разделение за счёт различного заряда разделяемых молекул д. Сорбция и десорбция Правильные ответы: 1-д, 2-а, 3-б, 4-г, 5-в.
5. К плоскостной хроматографии относятся:
а. Тонкослойная хроматография б. Газо-жидкостная хроматография в. Сверхвысокоэффективная жидкостная хроматография г. Высокоэффективная жидкостная хроматография д. Бумажная хроматография Правильные ответы: а, д.
6. К колоночной хроматографии относятся:
а. Тонкослойная хроматография б. Газо-жидкостная хроматография в. Сверхвысокоэффективная жидкостная хроматография г. Высокоэффективная жидкостная хроматография д. Бумажная хроматография Правильные ответы: б, в, г.
7. Обозначьте детали на приведённой ниже блок-схеме газового хроматографа:
а. Инжектор б. Термостат в. Колонка г. Детектор д. Интегратор е. Преобразователь сигналов ж. Ёмкость с газом-носителем Правильные ответы: 1-ж, 2-а, 3-в, 4-б,5-г, 6-е, 7-д.
8. В газовой хроматографии применяются следующие типы колонок:
а. Насадочные б. Ионообменные в. Капиллярные г. Металлические Правильные ответы: а, в.
9. Выберите газы, которые не используются в газовой хроматографии в качестве подвижной фазы:
а. Гелий б. Ксенон в. Кислород г. Азот д. Метан е. Ацетилен ж. Аргон Правильные ответы: в, д, е.
10. Выберите газы, которые могут использоваться в газовой хроматографии в качестве подвижной фазы:
а. Гелий б. Ксенон в. Кислород г. Азот д. Метан е. Ацетилен ж. Аргон Правильные ответы: а, б, г, ж.
11. Выберите типы детекторов, применяемых в газовой хроматографии:
а. Пламенно-ионизационный детектор б. Детектор по светорассеянию в. УФ-спектрофотометрический детектор г. Кондуктометрический детектор д. Детектор по теплопроводности е. Электронозахватный детектор ж. Масс-селективные детекторы з. Полярографический детектор Правильные ответы: а, д, е, ж.
12. На измерении степени силы тока в плазме пламени при сгорании веществ в токе водорода основан принцип действия:
а. Фотоионизационного детектора б. Детектора по теплопроводности в. Пламенно-ионизационного детектора г. Электрохимического детектора д. Амперометрического детектора Правильные ответы: в.
13. Методом газовой хроматографии можно разделять вещества:
а. Газообразные б. Летучие в. Водные растворы г. Термостабильные д. Термолабильные Правильные ответы: а, б, г.
14. В зависимости от полярности подвижной и неподвижной фаз в методе ВЭЖХ выделяют следующие подвиды:
а. Нормально-фазовая хроматография б. Ионообменная хроматография в. Распределительная хроматография г. Адсорбционная хроматография д. Обращённо-фазовая хроматография Правильные ответы: а, д.
15. В качестве подвижной фазы в обращённо-фазовой ВЭЖХ используют:
а. Метанол б. Гексан в. Толуол г. Ацетонитрил д. Этилацетат е. Изопропанол ж. Буферные растворы Правильные ответы: а, г, е, ж.
16. В качестве подвижной фазы в нормально-фазовой ВЭЖХ используют:
а. Метанол б. Гексан в. Толуол г. Ацетонитрил д. Этилацетат е. Изопропанол ж. Буферные растворы Правильные ответы: а, б, в, г, д, е.
17. К селективным детекторам в ВЭЖХ относятся:
а. Флуориметрический б. Масс-селективный в. Рефрактометрический г. Кондуктометрический д. Амперометрический е. УФ-спектрофотометрический ж. Фотодиодноматричный Правильные ответы: б, ж.
18. К неселективным детекторам в ВЭЖХ относятся:
а. Флуориметрический б. Масс-селективный в. Рефрактометрический г. Кондуктометрический д. Амперометрический е. УФ-спектрофотометрический ж. Фотодиодноматричный Правильные ответы: а, в, г, д, е.
19. Основные практические отличия УФ-спектрофотометрического детектора в ВЭЖХ от фотодиодноматричного:
а. Возможность измерять испускание света б. Возможность регистрации сигнала при нескольких длинах волн в. Возожность измерять светорассеяние г. Возможность регистрации спектра поглощения разделяемых веществ д. Более высокая чувствительность Правильные ответы: б, г.
20. Газовая хроматография в фармацевтическом анализе не применяется для:
а. Анализа подлинности б. Определения специфических примесей в. Количественного определения г. Разделения анализируемой смеси с целью проведения дальнейшего анализа д. Анализа остаточных органических растворителей Правильные ответы: г
21. Высокоэффективная жидкостная хроматография в фармацевтическом анализе применяется для:
а. Анализа подлинности б. Количественного определения в. Анализа чистоты г. Биоаналитических исследований д. Токсикологических исследований Правильные ответы: а, б, в, г, д.
22. При использовании масс-селективных детекторов в жидкостной хроматографии применяются следующие способы ионизации:
а. Электроспрей б. Электронная ионизация в. Химическая ионизация г. Направленный электронный удар д. Термоспрей Правильные ответы: а, д.
23. При использовании масс-селективных детекторов в газовой хроматографии применяются следующие способы ионизации:
а. Электроспрей б. Электронная ионизация в. Химическая ионизация г. Направленный электронный удар д. Термоспрей Правильные ответы: б, в, г.
24. В масс-селективных детекторах в газовой и жидкостной хроматографии используются следующие анализаторы:
а. Электроспрей б. Ионная ловушка в. Квадруполь г. Электронзахватный д. Времяпролётный е. Тройной квадруполь Правильные ответы: б, в, д, е.
25. Величина, характеризующая количество повторяемых взаимодействий компонентов разделяемой смеси с неподвижной фазой называется:
а. Высота эквивалентная теоретической тарелке б. Фактор ассиметрии в. Фактор симметрии г. Фактор разделения д. Число теоретических тарелок е. Индекс Ковача Правильные ответы: д.
26. Величина, характеризующая длину участка колонки, на который приходится один акт взаимодействия компонента разделяемой смеси с неподвижной фазой называется:
а. Высота эквивалентная теоретической тарелке б. Фактор ассиметрии в. Фактор симметрии г. Фактор разделения д. Число теоретических тарелок е. Индекс Ковача Правильные ответы: а.
27. Метод хроматографии был изобретён:
а. М. В. Ломоносовым б. А. И. Несмеяновым в. М. С. Цветом г. А. Эйнштейном д. А. Мартином и М. Сингом Правильные ответы: в.
28. Безразмерная величина, характеризующая удерживание вещества и равная отношению абсолютного объема удерживания к свободному объему колонки, называется:
а. Высота эквивалентная теоретической тарелке б. Фактор ассиметрии в. Фактор удерживания г. Фактор разделения д. Число теоретических тарелок е. Индекс Ковача Правильные ответы: в.
29. Безразмерная величина, характеризующая разделительную способность колонки по отношению к веществам А и Б и численно равная отношению факторов удерживания или приведенных времен (объемов) удерживания, называется:
а. Высота эквивалентная теоретической тарелке б. Коэффициент селективности в. Фактор удерживания г. Фактор разделения д. Число теоретических тарелок е. Индекс Ковача Правильные ответы: б
30. Время от момента ввода пробы вещества в хроматограф до момента регистрации максимума соответствующего хроматографического пика, называется:
а. Исправленное (приведённое) время удерживания б. Мёртвое время в. Абсолютное время удерживания Правильные ответы: в.
31. Время от момента ввода пробы несорбируемого вещества в хроматограф до момента регистрации максимума сигнала детектора, называется:
а. Исправленное (приведённое) время удерживания б. Мёртвое время в. Абсолютное время удерживания Правильные ответы: б.
32. Абсолютное время удерживания за вычетом мертвого времени, называется:
а. Исправленное (приведённое) время удерживания б. Мёртвое время в. Абсолютное время удерживания Правильные ответы: а.
33. Хроматография – это процесс:
А. Разделения смесей веществ, основанный на химическом взаимодействии разделяемых компонентов со второй контактирующей фазой.
Б. Разделения смесей веществ, основанный на количественных различиях в поведении разделяемых компонентов при их непрерывном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения.
В. Разделения смесей веществ, основанный на необратимом смешивании разделяемых компонентов во второй контактирующей фазе.
34. Хроматографический метод анализа является методом А. Качественного анализа Б.Количественного анализа В. И качественного, и количественного анализа В
35. Хроматографический метод анализа является А. Физическим методом анализа Б.Физико-химическим методом анализа В.Химическим методом аналиаза Б
36. Какого вида хроматографии не существует?
А.Тонкослойная Б. Ионобменная В.Потенциометрическая Г. Газожидкостная В
37. Способность вещества вращать плоскость поляризации при прохождении через него поляризованного света называется А. Оптической плотностью;
38. Оптический метод анализа, основанный на измерении угла вращения поляризованного света, называется – А. Рефрактометрия;
39. Метод поляриметрии основан на измерении А. Оптической плотности;
40. Удельное вращение не зависит от:
41. Атом углерода, у которого все заместители разные, называют А. Третичным;
42. Формула расчёта удельного вращения А. [] = ·100/l·C Б. С = ·100/[]·l В. C = n-n0/F Г. [] = ·t/l·C А
45. Формула расчёта концентрации оптически активного вещества в растворе А. [] = ·100/l·C Б. С = ·100/[]·l В. C = n-n0/F Г. [] = ·t/l·C Б
46. Вещества, способные изменять плоскость вращения поляризованного светаназывают А. Оптически вращающими;
47. Удельное вращение для оптически активного вещества это величина:
48. Удельное вращение измеряется в А. Градусах;
А.Отношение скорости распространения света в вакууме к скорости распространения света виспытуемым веществе Б.Величина отклонения плоскости поляризации от начального положения, выраженная в условных градусах В.Фактор, равный величине прироста показателя преломления при увеличении концентрации на 1% Б
50.Величина угла вращения не зависит от:
А.Природы оптически активного вещества Б.Длины пути поляризованного света В.Длины волны света Г.Природы растворителя Д.Плоскости поляризации света Е.Концентрации оптически активного вещества Е
51.Угол поворота плоскости поляризации монохроматического света на пути длиной в 1 дм в среде, содержащей оптически активное вещество, при условном приведении концентрации этого вещества к значению, равному 1 г/мл:
А.Угол вращения Б.Показатель преломления В.Удельное вращение Г.Показатель поглощения В
52.Концентрацию оптически активного вещества в растворе при использовании метода поляриметрии находят по формуле:
53. Формула для расчета процентного содержания используется в:
54. Методы расчета концентрации в рефрактометрии:
А. с использованием расчетной формулы.
Б. с использованием коэффициента Стьюдента В. с использованием специальных таблиц.
Г. с использованием титриметрического фактора пересчета.
55. Рефрактометрию используют для анализа:
А. только однокомпонентных растворов Б. только многокомпонентных растворов В. Однокомпонентных и многокомпонентных растворов Г. Ни одного из перечисленных В
56. Рефрактометрию для многокомпонентных растворов используют:
А. после предварительного определения концентрации одного из веществ Б. без предварительного определения концентрации одного из веществ В. Не используют вообще А
57. Точность измеренияпоказателя преломления по ГФ должна быть не ниже:
А. ± 110-4 Б. ± 210-4 В. ± 210-5 Г. ± 510-4 Б
58.Диапазонизмеряемыхпоказателейпреломленияприизмерениивпроходящемсвете по ГФ:
А. 1,3-1,7 Б. 1,0 – 1,7 В. 1,3 – 1,88 Г. 1,0 – 1,88 А
59. Рефрактометрическое определение по ГФ проводятпри:
А. температуре (20±0,5)С Б. плотности 1,05 – 1,88 В.длиневолнылинииDспектра натрия Г.длиневолнылинииDспектра калия А.В
60. Фактор F в формуле для расчетов при рефрактометрическом определении соответствует:
А. величинеприростапоказателяпреломленияпри увеличенииконцентрации на 1 % Б. величинеприростапоказателяпреломленияпри увеличенииконцентрации на 0,1 % В. Отношению показателей преломления растворителя и раствора Г. Разности показателей преломления растворителя и раствора вещества А
61. Зависимость показателя преломления от концентрации может быть:
А. только линейной Б. только нелинейной В. Как линейной так и нелинейной Г. Только обратной В
62. Абсолютным показателемпреломления(индексомрефракции)называют А.отношение скорости света в воздухе к скорости света в испытуемом веществе Б.отношение скоростисветаввакуумекскоростисветависпытуемомвеществе В. Отношение скоростисветависпытуемомвеществекскоростисветав вакууме Г. Отношение скорости света в испытуемом веществек скорости света в воздухе Б
63. Относительным показателемпреломления(индексомрефракции)называют А.отношениескорости света в воздухе к скорости света в испытуемом веществе Б.
отношениескоростисветаввакуумекскоростисветависпытуемомвеществе В. отношениескоростисветависпытуемомвеществекскоростисветав вакууме Г. отношениескорости света в испытуемом веществек скорости света в воздухе А
64. Согласно действующей ФС на р-р глюкозы для инъекций количественное определение проводят:
А. методом рефрактометрии Б. методом ВЭЖХ В. Методом поляриметрии Г. методом обратноййдометрии Г
65. Нижняя шкала в рефрактометре (в %) соответствует концентрации:
А. сахарозы Б. глюкозы В. молочного сахара Г. Этилового спирта Д. глицерина Б
66. Расчеты по рефрактометрическим таблицам ведется с использованием:
А. корреляции Б. экстраполяции В. поправочного коэффициента Г. коэффициента Стьюдента Б
67.Разновидностью рефрактометрии бывают методы:
а. Рефракто-денсиметрический б. рефракто-экстракционный в. Рефракто-полярографический г. рефракто- эмиссионный А,Б
68. Анализ спирто – водных растворов с концентрацией более 50% проводится:
а. с использованием разбавления б. с определением плотности в. с использованием поправочного коэффициента г. не проводится данным методом А
69. При анализе спирто – водных растворов используется:
а. Фактор пересчета б. Поправочный коэффициент концентрации в. Поправочный температурный коэффициент г. Поправочный парциальный коэффициент А,Б
70. Метод рефрактометрии используется для:
а водных растворов б. спиртовых растворов в. порошков г. таблеток А.Б.В
71. При анализе многокомпонентных порошков используют:
а. рефракто-титриметрический анализ б. системы линейных уравнений с двумя неизвестными в. рефракто-гравиметрический анализ г. формулы пересчета по массе А.Б
72. Для определения показателя преломления газов применяют:
а. поляриметр б интерферометр в. пикнометр г. газоанализатор Б
73. Показатель преломления зависит от факторов:
а. природы вещества б. плотности вещества в. температуры и давления, при которых проводится измерение г. длины волны света А.Б.В
74.Отношение скорости распространения света в вакууме к скорости распространения света виспытуемым веществе – это:
А.Оптическое вращение Б.Показатель преломления В.Показатель поглощения Г.Хроматография Б
75.Показатель преломления не зависит от:
А.Температуры Б.Давления В.Концентрации вещества Г.Природы растворителя Д.Длины волны света, при котором проводят определение
А.Концентрация раствора Б.Показатель преломления раствора В.Показатель преломления растворителя Г.Фактор, равный величине прироста показателя преломления при увеличении концентрации на 1% Г
77.Приборы, применяемые для определения показателя преломления, называются:
А. СпектрофотометрамиБ. ДенсиметрамиВ. РефрактометрамиГ.Хроматографами
78. В ультрафиолетовом спектрофотометре дифракционная решётка выполняет роль:
А.Светофильтра Б.Монохроматора В. Детектора Г. Источника света Б
79. Анализ веществ в растворах методом спектрофотометрии в видимой области спектра основан на способности веществ:
А.Испускать излучение Б.Поглощать свет в области длин волн от 380 до 780 нм В.Изменять плоскость вращения плоскополяризованного света Г.Поглощать свет в области длин волн от 190 до 380 нм Б
80. Величина оптической плотности не зависит от:
А.Структуры вещества Б.Концентрации вещества в растворе В.Плотности раствора Г.Толщины слоя В
81. В качестве раствора сравнения в методе спектрофотометрии ГФ XII рекомендовано использовать:
A. Смесь воды и спирта в соотношении 1:1
Б. Смесь раствора ванилина в спирте и раствора хлористоводородной кислоты B. Растворитель, или смесь растворителей, в которой растворено вещество Г. Воду В
82. Величина А1%1см называется:
A. Молярным показателем поглощения Б.Оптической плотностью B. Удельным показателем поглощения Г. Пропусканием В
83. Величину, представляющую собой оптическую плотность раствора вещества с концентрацией 10 г/л (1 г/100 мл) в кювете с толщиной слоя 1 см, ГФ XII определяет как:
A. Пропускание Б. Интенсивность падающего монохроматического излучения B. Молярный показатель поглощения Г. Удельный показатель поглощения Г
84. Закон Бугера-Ламберта-Бэра количественно описывает:
А. Уменьшение величины монохроматического излучения, проходящего через гомогенную поглощающую среду Б. Соотношение молярного и удельного показателя поглощения В. Изменение плоскости вращения плоскополяризованного света Г. Изменение показателя преломления в зависимости от концентрации А
85. Величина, определяемая как log10(1/Т), где Т – пропускание, называется:
А. Удельный показатель поглощения Б. Удельный показатель преломления В. Угол вращения Г. Оптическая плотность Г
86. В законе Бугера-Ламберта-Бэра символом обозначается:
А. Молярный показатель поглощения Б. Оптическая плотность В. Толщина кюветы Г. Удельный показатель поглощения А
87. Данная блок-схема отражает строение А. Фотоэлектроколориметра Б. Спектрофотометра В. Жидкостного хроматографа Г. Поляриметра Д. Рефрактометра Б
88. Метод спектрофотометрии основан на А. способности растворов веществ поглощать монохроматический свет Б. способности растворов веществ преломлять свет В. явлении сорбции-десорбции Г.способность растворов веществ вращать плоскость поляризации Д. способности вещества в возбужденном состоянии испускать свет А
89. Сульфацил натрия: можно определить спектрофотометрически в видимой области спектра по продуктам реакции с:
А. раствором гидроксидом натрия при нагревании Б. спиртовым раствором ванилина В. с нитритом натрия, а затем с -нафтолом Г. с калия йодидом Д. п-диметиламинбензальдегидом Б,В,Д
90. Измерение оптической плотности проводят:
А. в кювете с толщиной слоя 1 см Б. при охлаждении до 10С В. с добавлением стабилизаторов Г. при температуре 20±1С Д. с добавлением гольмия оксида А,Г
91. Концентрацию испытуемого раствора методом спектрофотометрии можно определить :
A. по калибровочному графику Б. по объему стандартного раствора В. по стандартному раствору Г. по закону Бугера-Ламберта-Бера Д. по интенсивности рассеянного света А,В,Г
92. Для снижения величины ошибки при определении оптической плотности испытуемого раствора:
А. Пробу термостатируют Б. концентрацию подбирают таким образом, чтобы значение плотности находилось в пределах 0,2 – 0,8 В. прибор калибруют Г. в прибор встраивают детектор Д. строят калибровочный график
93. В основе спектрофотометрии лежит объединенный закон Бугера-Ламберта-Бера, имеющий вид:
94.Спектрофотометры позволяют проводить анализ А. Бесцветных соединений в ультрафиолетовой и инфракрасной областях спектра Б. Окрашенных соединений в видимой области спектра В. Бесцветных и окрашенных соединений в видимой, ультрафиолетовой и инфракрасной областях спектра В.
95.Концентрацию вещества при спектрофотометрическом анализе находят по формуле:
96. Спектрофотометрия является методом:
А. Качественного анализа Б. Количественного анализа В. И качественного, и количественного анализа В
97. Способ разделения смесей, основанный на различном сродстве компонентов смеси к двум взаимно несмешивающимся фазам называется А. Рефрактометрией;
98. По расположению неподвижной фазы тонкослойная хроматография относится к:
99. По типу взаимодействия компонентов смеси и неподвижной фазы тонкослойная хроматография относится к:
А. Коэффициентом поглощения;
Б. Скоростью потока элюента;
В. Коэффициентом удерживания;
101. В роли подвижной фазы в методе ТСХ обычно выступает А. Вода;
Б. Система органических растворителей;
В. Растворы минеральных кислот;
Г. Растворы неорганических солей.
102. Тонкослойная хроматография выполняется на А. Хроматографической бумаге;
В. Жидкостном хроматографе;
Г. Хроматографической пластинке с нанесённым слоем сорбента.
103. Необходимым условием для проведения анализа методом ТСХ является:
А. Насыщение хроматографической камеры парами растворителя;
Б. Добавление в подвижную фазу соляной кислоты;
В. Использование в качестве неподвижной фазы силикагеля;
Г. Использования УФ-облучения в качестве детектора.
104. Коэффициент удерживания определяется как:
А. Расстояние от линии старта до линии финиша;
Б. Отношение расстояния от линии старта до центра пятна вещества к расстоянию от линии старта до линии финиша;
В. Отношение расстояния от линии старта до центра пятна определяемого вещества к расстоянию от линии старта до центра пятна стандарта;
Г. Отношение расстояния от линии старта до центра пятна к времени хроматографирования.
105. Расположите этапы анализа образца препарата методом ТСХ в порядке их проведения:
Б. Нанесение образца на хроматографическую пластину;
В. Приготовление неподвижной фазы и насыщение хроматографической камеры её парами;
106. Элюирование прекращают А. По истечению времени, указанного в методике;
Б. При достижении фронтом подвижной фазы конца пластинки;
В. При прохождении фронтом подвижной фазы линии финиша;
Г. При высыхании пластинки.
107. Раствор стандартного образца в методе ТСХ наносят на:
108. Величина Rsрассчитывается как:
109. В фармацевтическом анализе метод ТСХ не используетсядля:
А. Качественного анализа веществ;
Б. Количественного анализа веществ;
В. Определения чистоты препарата;
Г. Разделения смеси на компоненты.
110. Реактивом для обнаружения на хроматограмме алкалоидов является:
Б. Реактив Марки В. Бромфеноловый синий;
111. Тонкослойная хроматография основана на А. Процессе, протекающем при движении подвижной фазы в тонком слое сорбента (носителя), нанесенном на инертную поверхность Б. Процессе, протекающем на фильтровальной бумаге при перемещении по ее капиллярам и поверхности подвижной жидкой фазы В. Обратимой хемосорбции ионов анализируемого раствора ионогенными группами сорбента А
112. При проведении ТСХ, на дно хроматографической камеры наливают подвижной фазы в количестве А. 4-5 см Б. Половины объема от камеры В. Подвижную фазу льют на саму пластинку Г. 0,5-1 см Г
113. Для чего нужно насыщать хроматографическую камеру подвижной фазой при проведении ТСХ?
А. Для увеличения скорости передвижения веществ по пластинке Б. Для более точного результата исследования В. Для более полного разделения веществ А
114. Каким образом помещают пластинку с нанесенными веществами в хроматографическую камеру при проведении ТСХ?
А. Строго вертикально Б. Под углом 60-90 градусов В. Строго горизонтально Б
115. Что является результатом проведения ТСХ?
А. Пластинка с видимыми следами прохождения веществ по слою сорбента Б. Пластинка с видимыми пятнами веществ на слое сорбента В. Пластинка с видимыми или невидимыми пятнами веществ на слое сорбента, которые нужно детектировать согласно общей фармакопейной статье Г. Пластинка с видимыми или невидимыми пятнами веществ на слое сорбента, которые нужно детектировать согласно частной фармакопейной статье на анализируемый препарат Г
116. Положение зоны вещества на хроматограмме характеризуется величиной Rf, которая равна А. Отношению расстояния от одного пятна к другому к расстоянию от линии старта до линии финиша Б. Отношению расстояния от линии старта до линии финиша к расстоянию от стартовой линии до центра зоны вещества В. Отношению расстояния от стартовой линии до центра зоны вещества к расстоянию от стартовой линии до линии фронта.
117. Какие физические методы используются в анализе лекарственных веществ?
А. Определение температуры плавления Б. Определение температуры разложения В. Определение температуры затвердевания Г. Определение плотности А,В,Г
118. Какие из указанных методов анализа не относятся к физическим?
А. Определение плотности Б. Определение температуры кипения В. Хроматография Г. Определение вязкости В
119. Что подразумевают под термином «температура плавления»?
А. Температура начала плавления вещества Б. Интервал между началом и концом плавления вещества В. Температура конца плавления вещества Б
120. Как визуально определить начало и конец плавления вещества?
А. Невозможно, необходим инструментальный анализ Б. Начало плавления – переход половины массы вещества в жидкое состояние, конец – полный переход вещества в жидкое состояние В. Начало плавления – появление первой капли жидкости, конец – полный переход вещества в жидкое состояние
В121. Интервал между началом и концом плавления не должен превышать —
А. 2°С Б. 3°С В. 5°С Г. не регламентируется А
122. Какие методы используются для определения температуры веществ,
1) легко превращаемых в порошок и неустойчивых при нагревании
2) легко превращаемых в порошок и устойчивых при нагревании
3) для веществ, не растираемых в порошки а) 1 б) 1а в) 2 г) 3 1-Б, 2-А, 3 -В.Г
123. Температура затвердевания – А. Наиболее высокая, остающаяся в течение короткого времени постоянной температура во время перехода вещества из жидкого состояния в твердое Б. Наименее высокая, остающаяся в течение продолжительного времени постоянной температура во время перехода вещества из жидкого состояния в твердое В. Наиболее высокая, остающаяся в течение короткого времени постоянной температура во время перехода вещества из твердого состояния в жидкое А
1. Определение температуры пределов перегонки –
125. Определение вязкости – А. Основано на измерении вязкости с помощью погружения ареометра Б. Основано на измерении количества капель жидкости в определенном объеме с помощью вискозиметра В. Основано на сравнении числа капель исследуемой жидкости с числом капель воды очищенной в одном и том же объеме с помощью вискозиметра Б
Отметьте один или несколько правильных ответов:
126. К чему приводит превышение содержания влаги в лекарственных средствах:
А. Разложение активных молекул Б. Потеря фармакологической активности В. Появление токсических эффектов Г. Все вышеперечисленное верно Г
127. Отметьте цифрами верные и неверные высказывания относительно метода К.
А. Применим для малых навесок субстанций 1. Верное высказывание Б. Легкая пробоподготовка 2. Неверное высказывание В. Малое время анализа Г. Наиболее точно определяется содержание влаги в субстанциях, реагирующих с реактивом К. Фишера (сульфиды, меркаптаны и т.д.) Д. Пригоден для автоматизации Е. Можно анализировать твердые вещества, жидкости и газы Ж. Можно проводить без контрольного опыта А-1, Б-1, В-1, Г-2,Д-1, Е-1, Ж-1
128. Компоненты, входящие в состав реактива Фишера:
А. I2 Б. Безводный пиридин В. HCl Г. Абсолютный метанол Д. SO2 А.Б.Г.Д
129. Наиболее важная особенность реактивов, применяемых в методе К. Фишера:
А. Высокая токсичность Б. Высокая гигроскопичность В. Высокая стоимость Д. Стабильность при длительном хранении Б
130. Определение конца титрования по методу К. Фишера возможно:
А. Электрометрически Б. По изменению окраски индикатора тропеолина 00 В. По изменению окраски раствора от желтой до красновато-коричневой Г. По пожелтению осадка на дне колбы А.В.
131. Какую консистенцию при комнатной температуре образуют триглицериды, состоящие из насыщенных жирных кислот:
132. Какую консистенцию при комнатной температуре образуют триглицериды, состоящие из ненасыщенных жирных кислот:
133. Сопоставьте определение и понятие:
А. Количество миллиграммов едкого кали, необходимое для нейтрализации свободных кислот, содержащихся в 1 г исследуемого вещества Б. Количество миллиграммов едкого кали, необходимое для нейтрализации свободных кислот и кислот, образующихся при полном гидролизе сложных эфиров, содержащихся в 1 г исследуемого вещества.
134. Определение кислотного числа:
А. Прямое титрование Б Обратное титрование В. Алкалиметрия Г. Ацидиметрия А.В
135. Определение кислотного числа:
А. Прямое титрование Б Обратное титрование В. Алкалиметрия Г. Ацидиметрия Б,Г
136. Сопоставьте методику и особенность определения:
А. Определение кислотного числа 1. С контрольным опытом Б. Определение числа омыления 2. Без контрольного опыта А-2, Б-1
137. Отметьте верные высказывания относительно определения аминного азота методом формольного титрования:
А. Предварительно проводят реакцию аминных групп с формальдегидом Б. Высокая точность метода В. Прямая алкалиметрия Г. Обратная ацидиметрия А,В,Г
138. Отметьте верные высказывания относительно определения аминного азота методом йодометрического титрования:
А. Возможно определение для любых аминокислот Б. Возможно определение для серосодержащих аминокислот В. Прямая йодометрия Г. Обратное титрование с контрольным опытом А,Б,Г
139. Испытание на стерильность проводят для
140. Испытание на микробиологическую чистоту проводят для
Д) порошков для внутреннего применения Г,Д
141. Испытание на пирогенность проводят для
142. Испытание на аномальную токсичность проводят на
Д) тест-культурах микроорганизмов А,Б
143. Испытание на пирогенность проводят на
Д) тест-культурах микроорганизмов В
144. Показатель, который косвенно отражает возможную пирогенность препарата
Б) микробиологическая чистота
В) бактериальные эндотоксины
Д) гистамин и вещества гистаминоподобного действия В
145. Возможные методы проведения испытания на бактериальные эндотоксины
Б) турбидиметрическая методика
Д) измерение температуры тела кроликов А,Б,Г
146. Возможные методы количественного определения активности антибиотиков
Б) турбидиметрическая методика
Д) измерение температуры тела кроликов Б,В
147. Возможные методы определения пирогенности
Б) турбидиметрическая методика
Д) измерение температуры тела кроликов Д
148. Возможные методы количественного определения биологической активности
А) методы с использованием меченого антигена
Б) метод иммунопреципитации
Д) гель-хроматография А,Б,В,Г Химические методы анализа
1. Укажите фактор эквивалентности йода по реакции с натрия тиосульфатом?
2. Какой фактор эквивалентности имеет натрия тиосульфат по реакции с йодом?
3. Какой фактор эквивалентности имеет глюкоза по реакции с натрия гипойодитом?
4. Укажите фактор эквивалентности натрия гипойодита по реакции с натрия йодидом?
5. Какой фактор эквивалентности имеет натрия гипойодит при реакции с глюкозой?
6. Какая условная запись концентрации соответствует названию «сантимолярный раствор»?
A. 1 М B. 0,1М C. 0,5Н D. 0,01 Н E. 0,01 М
7. Какая условная запись концентрации соответствует названию «сантимонормальный раствор»?
A. 1 М B. 0,1М C. 0,5Н D. 0,01 Н E. 0,01 М
8. Выберите концентрацию, соответствующую названию «пятидецинормальный раствор»?
A. 5 М B. 0,5М C. 0,5Н D. 0,05 Н E. 0,05 М
9. Какая условная запись концентрации соответствует названию «пятисантинормальный раствор»?
A. 5 М B. 0,5М C. 0,5Н D. 0,05 Н E. 0,05 М
10. Выберите концентрацию, соответствующую названию «децинормальный раствор»?
A. 1 Н B. 0,1М C. 0,1Н D. 0,01 Н E. 0,01 М
11. Какой способ титрования используется при йодометрическом определении глюкозы?
A. Прямой B. Обратный C. Заместительный D. Косвенный
12. Метод нитритометрического титрования относят к:
A. Кислотно-основным B. Осадительным C. Комплексиметрическим D. Окислительно-восстановительным
13. Для определения конечной точки титрования при нитритометрическом титровании, ГФ XII рекомендует использовать:
A. Индикатор тропеолин 00 B. Йодкрахмальную бумагу C. Индикатор нейтральный красный D. Потенциометрический способ
14. Методом нитритометрического титрования после предварительного восстановления можно определять вещества, содержащие:
A. Вторичную ароматическую аминогруппу B. Первичную алифатическую аминогруппу C. Первичную ароматическую аминогруппу D. Ароматическую нитрогруппу
15. Методом нитритометрического титрования можно определять вещества, содержащие:
A. Вторичную ароматическую аминогруппу B. Первичную алифатическую аминогруппу C. Первичную ароматическую аминогруппу D. Ароматическую нитрогруппу
16. Какой продукт не образуется в ходе реакции диазотирования:
A. Соль диазония B. Натрия хлорид C. Азокраситель D. Вода
17. Фактор эквивалентностистандартного раствора натрия нитрита в количественном определении сульфацила-натрия:
18. ГФ XIIне рекомендует метод нитритометрического титрования для определения:
A. Веществ, содержащих первичную ароматическую аминогруппу B. Гидразидов C. Веществ, содержащих первичную алифатическую аминогруппу D. Веществ, содержащих ароматическую нитрогруппу
19. Необходимость охлаждения реакционной смеси при нитритометрическом титровании обусловлена:
A. Нестойкостью раствора натрия нитрита B. Разрушением индикатора при комнатной температуре C. Нестойкостью образующегося продукта D. Нестойкостью определяемого вещества при комнатной температуре
20. При проведении нитритометрического титрования добавление хлористоводородной кислоты в реакционную смесь обусловлено:
A. Стабильностью образующегося продукта в солянокислой среде B. Образованием при взаимодействии с нитритом натрия азотистой кислоты как диазотирующего агента C. Условиями изменения окраски индикатора D. Растворимостью определяемого вещества
21. При проведении нитритометрического титрования добавление раствора калия бромида в реакционную смесь обусловлено:
A. Стабилизацией соли диазония B. Каталитической способностью C. Влиянием на окраску индикатора D. Созданием соответствующего рН раствора
22. К физико-химическим методам количественного определения относятся:
A. Спектрофотометрия B. Титрование C. Гравиметрия D. Фотоэлектроколориметрия
23. К химическим методам количественного определения относятся:
A. Нитритометрия B. Комплексиметрия C. Гравиметрия D. ВЭЖХ
24. Титриметрический метод используют для:
A. Количественного определения B. Качественного анализа C. Определения примесей D. Определения растворимости
25. Для кислотно-основного титрования используют титранты:
A. HCl B. NaNO2 C. KOH D. KMnO4
26. Методом кислотно-основного титрования можно определять:
A. глюкозу B. аскорбиновую кислоту C. аминалон D. бария сульфат
27. Для окислительно-восстановительного титрования используют стандартные растворы:
A. KMnO4 B. KBrO3 C. CuSO4 D. KI
28. Методом окислительно-восстановительного титрования можно определять:
A. глюкозу B. аскорбиновую кислоту C. натрия нитрит D. магния сульфат
29. Методом комплексонометрического титрования можно определять A. алюминия гидроксид B. соляную кислоту C. кальция лактат D. калия ацетат
30. Фактор эквивалентности при комплексонометрическом определении обычно равен:
40. Поправочный коэффициент – это A. отношение реально полученной концентрации титрованного раствора к теоретически заданной B. отношение теоретически заданной концентрации титрованного раствора к реально полученной C. отношение концентрации определяемого вещества к концентрации титранта D. отношение реально полученной концентрации определяемого раствора к теоретически заданной
41. Поправочный коэффициент для титрованного раствора вычисляют по формуле:
A. k= теорети е а B. k= а ти е а титрованного ра твора C. k= о редел е ого ве е тва о редел е ого ве е тва D. k= титрованного ра твора
42. Стандартные растворы хранят A. при комнатной температуре B. в прохладном темном месте C. в защищенном от солнечных лучей месте D. 3 года
43. Титрованные растворы стандартизуют A. по методикам Фармакопеи последнего издания B. титриметрическим методом C. физико-химическими методами
44. При приготовлении стандартных растворов для окислительновосстановительного титрования, имеющих меньшую, чем описано в ГФ, молярность:
A. нуждаются в переустановке поправочного коэффициента B. имеют такой же поправочный коэффициент C. готовят путем разбавления описанных в ГФ растворов D. готовят на воде, свободной от углерода диоксида
45. Согласно методике ГФ, поправочный коэффициент титрованного раствора натрия нитрита определяют по A. кислоте бензойной B. калия гидрофталату C. кислоте сульфаниловой D. кислоте сульфаминовой
46. Согласно методике ГФ, поправочный коэффициент титрованного раствора натрия тиосульфата определяют по A. Стандартному раствору йода B. Стандартному раствору калия бромата C. Стандартномураствору натрия нитрита D. кислоте аскорбиновой
47. Согласно методике ГФ, поправочный коэффициент титрованного раствора натрия гидроксида определяют по A. Стандартному растворукислоты серной B. Стандартному растворукислоты хлористоводородной C. кислоте салициловой D. кислоте сульфаниловой
48. Величина эквивалента в окислительно-восстановительном титровании соответствует A. количеству протонов, участвующих в реакции B. стехиометрическому коэффициенту перед определяемым веществом C. стехиометрическому коэффициенту перед титрантом D. количеству электронов, отданных или принятых определяемым веществом
49. Величина эквивалента в кислотно-основном титровании соответствует A. количеству протонов, участвующих в реакции B. стехиометрическому коэффициенту перед определяемым веществом С. по количеству молекул воды в уравнении реакции Д. количеству электронов, отданных или принятых определяемым веществом
50. Если поправочный коэффициент титрованного раствора равен 0,89, то раствор нужно A. укрепить более концентрированным раствором B. укрепить раствором лекарственного вещества C. разбавить водой D. разбавить менее концентрированным раствором
53. Для чего производят перекристаллизацию основных реактивов?
A. для растворения B. для очистки C. для концентрирования D. для установки титра
54. Протогенный растворитель, применяемый в кислотно-основном титровании в неводной среде A. ацетон B. ледяная уксусная кислота C. пиридин D. диметилформамид
55. Растворитель, применяющийся в кислотно-основном титрованиив неводной среде веществ, проявляющих основные свойства:
A. ацетон B. ледяная уксусная кислота C. пиридин D. диметилформамид
56. Протофильные растворители, применяемые в кислотно-основном титровании в неводной среде A. ацетон B. ледяная уксусная кислота C. пиридин D. диметилформамид
57. Растворитель, применяющийся в кислотно-основном титрованиив неводной среде веществ, проявляющих кислотные свойства:
A. ацетон B. ледяная уксусная кислота C. пиридин D. диметилформамид
58. Индикаторы, которые используют в комплексонометрии A. фенлфталеин B. ксиленоловый оранжевый C. эриохром черный D. метилоранж
59. Индикаторы, которые используют в кислотно-основном титровании A. крахмал B. фенолфталеин C. бромтимоловый синий D. кислотный хром темно-синий
60. Индикатор, которые не используют в комплексонометрии A. тропеолин 00 B. ксиленоловый оранжевый C. эриохром черный D. хальконкарбоновая кислота
61. Индикатор, который не используют в кислотно-основном титровании A. крахмал B. фенолфталеин C. бромтимоловый синий D. метиловый оранжевый
62. Фактор эквивалентности в алкалиметрическом определении борной кислоты равен A. 1 B. 3 C. 1/3 D. 1/2
63. Для усиления кислотных свойств борной кислоты при количественном определении добавляют:
A. фенолфталеин B. ледяную уксусную кислоту C. глицерин D. глицерин, нейтрализованный по фенолфталеину
64. Окислительно-восстановительным методом по ГФ определяют A. натрия нитрит B. кислоту аскорбиновую C. глутаминовую кислоту D. железа (II) сульфат
65. Для количественного определения веществ, обладающих восстановительными свойствами, могут быть использованы методы A. перманганатометрии B. цериметрии C. йодхлорметрии D. алкалиметрии
66. Для количественного определения веществ, обладающих окислительными свойствами, используют стандартный раствор A. натрия тиосульфата B. калия йодата C. хлорной кислоты D. аммония тиоционата
67. Для количественного определения веществ, обладающих кислотными свойствами, используют стандартный раствор A. калия перманганат B. калия гидроксид C. калия бромат D. калия йодид
68. Для количественного определения веществ, обладающих основными свойствами, используют стандартные растворы A. кислота хлористоводородная B. кислота хальконкарбоновая C. кислота хлорная D. кислота уксусная ледяная
69. Какое общее химическое свойство лекарственных средств: раствора водорода пероксида, натрия нитрит, железа (II) сульфат, натрия тиосульфат – позволяет использовать для их количественного определения окислительновосстановительную реакцию:
A. кислотные свойства B. основные свойства C. окислительные свойства D. восстановительные свойства
70. Для количественного анализа лекарственных средств, имеющих в молекуле первичную ароматическую аминогруппу, может быть использован метод A. ацидиметрии B. нитритометрии C. аргентометрии D. комплексонометрии E. алкалиметрии
71. Для цинка оксида, магния сульфата, висмута нитрата основного, кальция хлорида общим методом количественного определения является:
A. гравиметрия B. перманганатометрия C. йодометрия D. комплексонометрия E. нитритометрия
72. Внешним индикатором в нитритометрии является только один из перечисленных индикаторов:
A. кристаллический фиолетовый B. тропеолин 00 + метиленовая синь C. йод крахмальная бумага D. метиловый оранжевый + метиленовая синь E. универсальный индикатор
73. Какое из лекарственных веществ можно определять методом нитритоиетрии:
74. Количественное определение лекарственных веществ нитритометрическим методом требует соблюдения условий:
A. кислая среда B. ограничение скорости титрования C. пониженная температура реакционной смеси D. добавление калия бромида E. все выше перечисленное верно
75. Лекарственные средства определяемые количественно методом комплексонометрии:
A. натрия тиосульфат, калия хлорид, кальция хлорид B. натрия тиосульфат, калия хлорид, магния сульфат C. магния сульфат, цинка сульфат, кальция хлорид D. калия хлорид, кальция хлорид, магния сульфат E. магния сульфат, цинка сульфат, калия хлорид
76. Метод нитритометрии можно использовать для лекарственных веществ:
77.Метод количественного анализа в аналитической и фармацевтической химии, основанные на измерении объёма раствора реактива известной концентрации, расходуемого для реакции с определяемым веществом – это
78. Момент титрования, когда число эквивалентов добавляемого титранта равно числу эквивалентов определяемого вещества – A. Титриметрический фактор B. Теоретический объем титранта C. Точка эквивалентности
79. Какого вида титриметрического анализа не существует?
A. Кислотно-основное B. Окислительно-восстановительное C. Осадительное D. Нейтрализующее
A. к раствору определяемого вещества (аликвоте или навеске, титруемому веществу) добавляют небольшими порциями раствор титранта (рабочий раствор) B. к раствору определяемого вещества добавляют сначала заведомый избыток специального реагента и затем титруют его остаток, не вступивший в реакцию C. к раствору определяемого вещества добавляют сначала заведомый избыток специального реагента и затем титруют один из продуктов реакции между анализируемым веществом и добавленным реагентом
81. При обратном титровании:
A. к раствору определяемого вещества (аликвоте или навеске, титруемому веществу) добавляют небольшими порциями раствор титранта (рабочий раствор) B. к раствору определяемого вещества добавляют сначала заведомый избыток специального реагента и затем титруют его остаток, не вступивший в реакцию C. к раствору определяемого вещества добавляют сначала заведомый избыток специального реагента и затем титруют один из продуктов реакции между анализируемым веществом и добавленным реагентом
82. При заместительном титровании:
A. к раствору определяемого вещества (аликвоте или навеске, титруемому веществу) добавляют небольшими порциями раствор титранта (рабочий раствор) B. к раствору определяемого вещества добавляют сначала заведомый избыток специального реагента и затем титруют его остаток, не вступивший в реакцию C. к раствору определяемого вещества добавляют сначала заведомый избыток специального реагента и затем титруют один из продуктов реакции между анализируемым веществом и добавленным реагентом
83. Формула расчета концентрации при прямом титровании:
84. Формула расчета титриметрического фактора пересчета:
85. Формула расчета концентрации при обратном титровании:
86. Формула расчета концентрации при заместительном титровании:
87-91. Подбери соответствие при комплексонометрическом титровании по ГФ ХII « катион – используемый индикатор»:
87. катион алюминия (а) а) ксиленоловый оранжевый
88. катион магния (в) б) хромовый темно-синий
89. катион висмута (а) в) кислотный хром черный
90. катион цинка (а) г) хальконкарбоновая кислота
92. Окраска раствора в точке эквивалентности при комплексонометрическом титровании обусловлена образованием A. комплекса металла с ЭДТА B. комплекса металла с индикатором C. свободного индикатора D. комплекса металла с буферным раствором
93. Содержание лекарственного вещества в анализируемом образце расчитывают по формуле При использовании метода A. рефрактометрии B. поляриметрии C. спектрофотометрии D. хроматографии E. титриметрии
94. В комплексонометрическом титровании в качестве стандартного раствора используют раствор:
A. кислоты хлороводородной B. трилона Б C. тиосульфата натрия D. сульфат церия
95. Натрия эдетат образует с катионами различных металлов устойчивые и хорошо растворимые в воде комплексонаты металлов в стехиометрическом отношении:
96. Как называются индикаторы в комплексонометрическом титровании:
A. металлоиндикаторами B. адсорбционными C. редокс-индикаторы D.азоиндикаторы
97. Титрование с тропеолином 00 проводят до перехода окраски:
А. от красной к желтой B. от красно-фиолетовой к голубой C. от красной с голубой D. от синей к красно-фиолетовой
98. В качестве внутренних индикаторов при нитритометрическомтировании не используют:
А. тропеолин 00 B.тропеолин 00 в смеси с метиленовым синим C. нейтральный красный D.ксиленоловый оранжевый
99. При нитритометрическом титровании точку эквивалентности определяют:
A. электрометрическими методами B. с помощью внутренних индикаторов C. с помощью внешнего индикатора D. все выше перечисленное верно
100. При нитритомерическомтитровани титрование проводят при температуре раствора:
A. 15 20 С В. 18 20 С С. При комнатной температуре D. температура не имеет значения
106. Приготовленные титрованные растворы стандартизуют:
А. по стандартному титрованному раствору B. по точной навеске исходного стандартного вещества C. не стандартизуют
107. Концентрацию титрованных растворов определяют по общей статье «Титрованные растворы» путем достаточного количества титрований :
A. не менее 5 раз B. не менее 3 раз С. не менее 10 раз D. количество раз титрования не отражено в статье
 «КОМПЬЮТЕРНЫЕ НАУКИ И ТЕХНОЛОГИИ УДК 519.173 ИНТЕГРАЦИЯ СТРУКТУРИРОВАННЫХ ДАННЫХ В НЕОДНОРОДНЫХ ИНФОРМАЦИОННЫХ СИСТЕМАХ А.В. Чабей Кафедра ПМиИ ДонНТУ В статье описаны подходы автоматизации предприятия: комплексный, лос. »
«КОМПЬЮТЕРНЫЕ НАУКИ И ТЕХНОЛОГИИ УДК 519.173 ИНТЕГРАЦИЯ СТРУКТУРИРОВАННЫХ ДАННЫХ В НЕОДНОРОДНЫХ ИНФОРМАЦИОННЫХ СИСТЕМАХ А.В. Чабей Кафедра ПМиИ ДонНТУ В статье описаны подходы автоматизации предприятия: комплексный, лос. »
«AXMEд XYЛYCИ ^ АЛЛАХ B COOTBETCTBИИ C OБЂЯCHEHИЯMИ MOXAMMAдA AXMEд XYЛYCИ АЛЛАХ ^ CONTENTS ПРЕДИСЛОВИЕ К АНГЛИЙСКОМУ ПЕРЕВОДУ. 6 НАША КНИГА БЫЛА НАЗВАНА АЛЛАХ МОХАММЕДА 12 ОТ БОГА. »
«Белла Верникова Одесский текст: от Осипа Рабиновича к Юшкевичу и Жаботинскому* О заседаниях ЛАО писал в газете «Одесские новости» молодой журналист Владимир Жаботинский, сост. »
«Государственное бюджетное профессиональное образовательное учреждение Бирский многопрофильный профессиональный колледж Согласовано Утверждаю Методист зам.по УВР Л.А.Шкатова _Е.Н.Литвинова «»_2013г «»_2013г М. »
«Инв. № _ Томская федерация туризма Томский Клуб Туристов Отчет о горном походе 5 к. с. по Киргизскому Ала-тоо (Северный Тянь-Шань) совершенном с 1 по 17 августа 2003 г. Маршрутная книжка № O 67 03 Руководитель группы: Д.П. Стальмаков. Адрес р. »
«Введение в Python и Eric Иван Хахаев, 2009 Строки и последовательности В главе «Простой ввод и вывод» мы уже имели дело со строками (данными символьного типа). Теперь рассмотрим работу со строками в Python более детально. Строка представляет собой конечный набор символов. »
«Кругова И. Н.ТЕОРИЯ ЧЕЛОВЕЧЕСКОГО КАПИТАЛА: ПРЕДПОСЫЛКИ СОЗДАНИЯ, ЭВОЛЮЦИЯ РАЗВИТИЯ В РОССИИ Адрес статьи: www.gramota.net/materials/1/2008/9/37.html Статья опубликована в авторской редакции и от. »
«ПРОФЕССИОНАЛЬНОЕ НАВИГАЦИОННОЕ ПО “LIBELLE MAP” ВЕРСИЯ 2.1 РУКОВОДСТВО ПОЛЬЗОВАТЕЛЯ Оглавление Информация об авторских правах Предупреждения и информация о безопасности Краткие сведения о программе Наши партнеры : Установка программы Операционная система Android : Операционная система WinCE / Windows Mo. »
«Комплекс подготовки документов аэронавигационной информации Создание базы аэронавигационных данных Общие положения Основным источником аэронавигационной информации комплекса является реляционная база аэронавигационных данных. »
«Автоматизированная копия 586_306869 ВЫСШИЙ АРБИТРАЖНЫЙ СУД РОССИЙСКОЙ ФЕДЕРАЦИИ ПОСТАНОВЛЕНИЕ Президиума Высшего Арбитражного Суда Российской Федерации № 2929/11 Москва 6 сентября 2011 г. Президиум Высшего Арбитражного Суда Росси. »
«ПОЯСНЕНИЯ К ТАБЛИЦАМ С ЦЕЛЕВЫМИ ИНДИКАТОРАМИ И ПОКАЗАТЕЛЯМИ 1 ПРОГРАММЫ Число публикаций по результатам исследований и разработок в научных журналах, индексируемых в базе данных Scopus или в базе данных Сеть науки (WEB of Science) Под публикацией понимается письменный труд, доступный для м. »
«БОРИС РАББОТ: Шестидесятник, которого не услышали BORIS RABBOT: An Unheeded Voice of the 1960s A M E MOR I AL VOLU ME Boris Rabbot: An Unheeded Voice of the 1960s Articles Interviews Reminiscences Compiled by: Lynn Visson Vasiliy Arkanov Moscow, 2012 «R.Valent» М ЕМ ОРИ А ЛЬН. »
2017 www.lib.knigi-x.ru — «Бесплатная электронная библиотека — электронные матриалы»
Материалы этого сайта размещены для ознакомления, все права принадлежат их авторам.
Если Вы не согласны с тем, что Ваш материал размещён на этом сайте, пожалуйста, напишите нам, мы в течении 1-2 рабочих дней удалим его.
Источник
Определение сульфидов колориметрическим методом основано на образовании метиленовой сини в результате взаимодействия сульфвд-ионов с диметилпарафенилендиамином в присутствии Ре в качестве окислителя. Оптическую плотность измеряют на фотоэлектрокопориметре Ф ЭК-56 со светофильтром № 9 в кювете длиной 20 мм. При содержании сульфидов 0,02-0,4 мг/л относительная ошибка определения составляет 2-20% . [c.34]
Выбор концентраций. Концентрация должна быть такой, чтобы оптическая плотность раствора находилась в пределах от 0,2 до 0,5. При указанных значениях оптической плотности относительная ошибка определения концентрации на всех типах приборов будет минимальной. [c.347]
Отсюда следует, что минимальная относительная ошибка определения концентрации будет отвечать оптической плотности О = 0,434 или 36,8 % пропускания. [c.651]
Зависимость величины относительной ошибки определения следов воды при оптимальных условиях для ряда растворителей показана на рис. 68. Интервал концентраций воды, допускающих определения по поглощению в области основных частот валентных колебаний ОН-групп при неизменной величине АО/О, ограничен, с одной стороны, точностью изготовления кювет (большие концентрации воды), с другой — собственным поглощением растворителя (малые концентрации воды). Наиболее благоприятным для большинства растворителей является интервал концентраций 0,1—1,0%. Градуировочные графики для этого интервала, построенные в координатах оптическая плотность — содержание воды в растворе, прямолинейны. Результаты определения спектрофотометрическими методами хорошо совпадают с данными дру- [c.156]
Через анализируемый соляно- или азотнокислый раствор, содержащий 0,5—10 мг/мл плутония, пропускают сернистый газ в течение 5—-10 мин. Раствор выдерживают 15—20 мин. для полного восстановления плутония до трех-валентного состояния. Концентрацию кислоты в растворе поддерживают равной 0,5—1,5 М. 2,5—3 мл восстановленного раствора помещают в кювету с толщиной слоя 10 мм и Измеряют оптическую плотность при 560 и 600 ммк. Концентрацию плутония рассчитывают по формуле (1). Относительная ошибка определения составляет 0,5%. [c.153]
Выбор оптимальных условий анализа проводится различными способами наиболее простой из них — предварительное построение серии калибровочных графиков. Концентрации препаратов в растворах сравнения подбирают таким образом, чтобы оптическая плотность отличалась на 0,2—0,4. На каждом из построенных графиков устанавливают величину относительной погрешности, используя анализируемые растворы с относительной оптической плотностью 0,4—0,5. Оптимальными считают те концентрации раствора сравнения и анализируемого раствора, с помощью которых достигнута наименьшая относительная ошибка определений. [c.41]
Для построения градуировочного графика в мерные колбы вместимостью 100 мл отбирают 10,0 11,0…15,0 мл раствора р,1 мг/мл Nb(V), добавляют до 15 мл 6 %-ного раствора винной кислоты, 6 мл концентрированной НС1 и 20 мл 0,15 %-ного раствора кислотного хром фиолетового К. Раствор нагревают 5—7 мин при 60—70 С и после охлаждения разбавляют до метки водой. Измеряют оптическую плотность при 570 нм в кювете 1 см по раствору, содержащему 1 мг Nb(V) в 50 мл. Относительная ошибка определения составляет 1 %. [c.115]
Концентрацию разбавленного раствора пробы антрацена готовят так, чтобы оптическая плотность на максимумах находилась в пределах 0,5—0,8, так как относительная ошибка определения концентрации будет минимальная при этих значениях. [c.372]
Дифференциальный метод [11] следует применять при высоком содержании определяемого компонента в анализируемом материале. В дифференциальном методе оптическую плотность исследуемого раствора измеряют по отношению не к чистому растворителю (или раствору реактивов), а к раствору, содержащему известное коли честно определяемого вещества. Относительная ошибка определения концентрации этим методом уменьшается с увеличением концентрации нулевого раствора и получается наименьшей, когда его оптическая плотность и плотность исследуемого раствора одинаковы. Рекомендуется применять нулевой раствор такой концентрации, чтобы значения оптической плотности, соответствующие разности концентраций растворов исследуемого и нулевого, лежали в оптимальной области измерений оптических плотностей (при работе на ФЭК-М с левым барабаном лучше проводить измерения в интервале 0,3—0,7 единицы оптической плотности). [c.11]
Теоретически и экспериментально доказано, что при значении оптической плотности Д = 0,434 (что соответствует светопропусканию 36,8%) ошибка измерения будет наименьшей. На рис. 27 показана зависимость относительной ошибки при определении концентрации растворов от величины измеряемой оптической плотности. Минимальная ошибка А ин =0 2,9 наблюдается в [c.63]
Как видно из рис. 3.2 и 3.3, относительная ошибка определения концентрации зависит не только от величины стандартного отклонения пропускания Sj, но и от пропускания (оптической плотности) раствора, т. е. от концентрации исследуемого раствора. [c.48]
Относительная ошибка определения концентрации дифференциальным методом уменьшается с увеличением концентрации Сд раствора сравнения и получается наименьшей, когда светопоглощение или оптическая плотность исследуемого раствора и раствора сравнения почти одинаковы (С . Ср и Г I 1). При оптимальных условиях, когда отклонения от основного закона светопоглощения ничтожно малы, точность дифференциального метода не уступает точности классических объемных методов. Область концентраций, где соблюдается основной закон светопоглощения, определяется обычным путем (стр. 19) по данным измерений оптических плотностей [c.105]
Относительная ошибка определения концентрации раствора будет различной при работе на разных участках шкалы прибора и достигает минимума при значении оптической плотности равной 0,4. Поэтому при работе на приборе рекомендуется путем соответствующего выбора кювет ра)ботать вблизи указанного значения оптической плотности раствора. Предварительный выбор кювет проводят визуально, соответственно интенсивности окраски раствора. Если раствор интенсивно окрашен (темный), то следует пользоваться (в соответствии с основным уравнением колориметрии) кюветой с малой рабочей длиной (1—3 мм). В случае слабо окрашенных растворов рекомендуется работать с кюветами с большей рабочей длиной (30—50 мм). При измерении ряда растворов кювету заполняют раствором средней концентрации. Если полученное значение оптической плотности составляет примерно [c.367]
На рис. 1 показаны найденные нами области практически полной экстракции ФМК и ММК из растворов различных кислот, имевших различную концентрацию и содержавших различные количества молибдена. Видно, что оптические плотности экстрактов, содержащих 1 мкг As, в 2—2,4 раза меньше, чем у экстрактов, содержащих 1 мкг Р, что, очевидно, вытекает из соотношения атомных весов мышьяка и фосфора. Из графиков на рис. 1 видно, что при концентрации молибденовокислого аммония 0,25% получаются узкие (по кислотности) интервалы полной экстракции ФМК (кривые 1—3), в связи с чем его концентрация была повышена до 0,5%. При этом наиболее широкие области полной экстракции фосфора и мышьяка имела азотнокислая среда. Однако опыты показали, что в азотнокислой среде получаются несколько заниженные результаты определения мышьяка. Поэтому для определения выбрали солянокислую среду в области концентраций 0,8—1 н., где достигается практически полная экстракция ФМК и ММК (кривые 4 и S). В таблице помещены результаты последовательного экстракционно-фотометрического определения Ы0 % Р и Ы0 % As в азотнокислом алюминии особой чистоты, выполненного по описанному ниже ходу анализа, а также результаты аналогичных определений с дополнительным введением в растворы навесок этих солей примесей фосфора и мышьяка. При этом получены удовлетворительные результаты. На одно определение уходит 1 часа. Относительная ошибка определения 20%. [c.182]
Если эта модификация не сказывается на погрешности прибора при измерении оптической плотности, использование эталона в качестве раствора сравнения переносит пропускание в середину шкалы, где ошибка прибора оказывает минимальное влияние на относительную ошибку определения концентрации. [c.155]
Все спектры изображены в виде графиков зависимости а = /(Я), где сс — удельный коэффициент погашения илн экстинкции (оптическая плотность раствора, содержащего 1 г л вещества, при толщине слоя 1 см) и Я — длина волны (ммк). Масштабы спектров различны и выбраны таким образом, чтобы все характерные особенности спектра были наиболее наглядны. Включенные в атлас спектры получены авторами в лабораториях ВНИИСКа на спектрофотометре СФ-4 при комнатной температуре (20 3°С). Многие из них получены впервые. Относительная ошибка определения, обычная для СФ-4, составляла до 5%. В некоторых случаях полученные авторами данные не совпадают с литературными (приводятся в сносках при таблицах). Следует отметить, что в ряде случаев приходилось иметь дело с техническими продуктами или с веществами, степень чистоты которых была неизвестна. Это относится в основном к антиоксидантам и различным ингредиентам, применяемы.м при синтезе каучуков. [c.4]
Поскольку прибор дает возможность измерять пропускание с точностью до 1% и при этом минимальная относительная ошибка определения оптической плотности составляет 2%, значения е и к имеет смысл вычислять только до третьей значащей цифры. [c.90]
Относительная ошибка определения концентрации дифференциальным методом уменьшается с увеличением концентрации q раствора сравнения и получается наименьшей, когда светопоглощение или оптическая плотность исследуемого раствора и раствора [c.108]
Чтобы найти оптическую плотность, при которой относительная ошибка определения минимальна, продифференцируем (11.41) по Т х при постоянных АГ и Гер и приравняем производную нулю [c.63]
Определение общего содержания фосфатов фотоколориметрическим методом основано на образовании устойчивого желтого комплекса состава Н,РО -HVOj-ИМо Oj-w Н О и измерении его оптической плотности относительно раствора сравнения, содержащего определенное количество фосфатов. Относительная ошибка определения фосфатов в удобрениях, содержащих до 70 % , составляет +1,0%. [c.138]
Подготовку и анализ образцов СКН проводят так же, как и эталонных образцов. Содержание НАК находят по градуировочному графику (также пересчитывая величину оптической плотности на концентрацию 0,01 г/мл). Относительная ошибка определения составляет 3%, продолжительность анализа 2,0—2,5ч. [c.21]
Линейная зависимость между оптической плотностью и содержанием рения соблюдается для концентраций ниже 25 мкг[мл. Средняя относительная ошибка определения рения составляет +1, 27%. Определению рения разработанным методом не мешают те элементы, которые не поглощают в ультрафиолетовой части спектра (в области 230—280 нм). Оценка мешающего влияния некоторых элементов приводится в табл. 2. [c.65]
В зависимости от предполагаемой концентрации триметилкарбинола в изобутилене берется от 100 до 500 мл пробы. Пользуясь градуировочной кривой, по найденному значению величины оптической плотности находят количество триметилкарбинола в пробе в весовых процентах. Оптическая плотность определяется по методу базисной линии. Чувствительность метода 1-10″ вес.%. Продолжительность определения около 2 час. Относительная ошибка определения +15%. [c.196]
Для субъективных методов относительная ошибка определения интенсивности пропущенного излучения остается постоянной в некотором диапазоне интенсивностей, вследствие чего опшбка определения концентрации оказывается обратно пропорциональ-лой оптической плотности. Практически, потеря способности раз-]решения яркостей при оптических плотностях, превышающих 1 [c.634]
Раствор, содержащий до 30 мкг НеО , переносят в делительную воронку па 50 мл, добавляют 0,6 мл 18 N Н2804, 1 мл 0,1 %-ного раствора реагента и бидистиллатом доводят объем до 12 мл. Добавляют 6 мл бензола и экстрагируют 1 мин. После разделения фаз экстракт центрифугируют и измеряют оптическую плотность при 610 нм в кювете с 1= I см относительно экстракта холостого опыта. Количество репия тгаходят по калибровочному графику. Относительная ошибка определения пе превышает +3%. [c.131]
Фотометрическое определение кобальта после экстракции 1-нитрозо-2-нафтолата четыреххлористым углеродом [1138]. Навеску почвы (или растительного материала) разлагают фтористоводородной кислотой после озоления. Затем окисляют двухвалентное железо 37о-ным раствором Н2О2 и осаждают его в виде фосфата из уксуснокислого раствора, содержащего мочевину. К фильтрату прибавляют щелочный раствор 1-нитрозо-2-нафтола и извлекают окрашенный комплекс кобальта четыреххлористым углеродом. Экстракты промывают последовательно концентрированной соляной кислотой, водой, смесью (1 1) этанола и 0,1 Л/ NaOH. Оптическую плотность объединенных экстрактов измеряют при 400 ммк. Содержание кобальта находят по калибровочному графику. Относительная ошибка определения 0,3—0,5- 10″ % Со достигает 5%. [c.212]
При исследовании смеси фенолов используют деформационные колебания связей СН. Для количественного анализа изомерных крезолов наиболее удобными являются полосы поглощения, отвечающие неплоским колебаниям группы СН ароматического кольца 756 см для о-крезола, 778 см- для -крезола и 816 см- -для п-крезола [109, ПО]. Спектр снимают в растворе сероуглерода при концентрации крезолов в пределах 1,5—5,0%, где выполняется закон Ламберта — Бера, с толщиной кюветы 0,06—0,1 мм. Ввиду симметричности полос и значительного их удаления друг от друга оптическую плотность можно определять по методу базисной линии. Количественный расчет проводят по калибровоч-, ным графикам. Относительная ошибка определения составляет 1%. Аналогично анализируют содержание ксиленолов, рекомендуемые аналитические частоты которых равны 815, 834, 995 и 1000 СМ- соответственно для 2,4- 3 5- 2,5- ш 3,4.-изомеров, [111] [c.57]
Теоретически и экспериментально доказано, что при значении оптической плотности О — 0,434 (что соответствует светопропуска-нию 36,8%) ошибка измерения будет наименьшей. На рис. 38 показана зависимость относительной ошибки при определении концентрации растворов от величины измеряемой оптической плотности. Минимальная ошибка Амии = 2,9% наблюдается в интервале 0,3—0,7 единиц оптической плотности при измерении меньших и больших оптических плотностей ошибки измерения возрастают измерения О = 0,1 и 1,3 уже производятся с ошибкой, равной 2А ин, т. е. в 5,8%. [c.57]
Величина Ас/с есть мера относительной ошибки при измерении, концентрации, обусловленной абсолютной ошибкой АТ измерения Т, или ///о. Читателю следует обратить внимание на то, что относительная ошибка определения концентрации является функцией величины Т. Эта зависимость иллюстрируется данными табл. 24-4, причем предполагается, что абсолютная ошибка Т для серии измерений пропускания равна 0,005, или 0,5% (по рекомендации Хиски [18], за АТ принимают удвоенное среднее отклонение повторных измерений пропускания Т одного раствора) относительная ошибка определения концентрации проходит через минимум при оптической плотности, равной примерно 0,4. [Приравняв производную выражения (24-5) нулю, можно показать, что этот минимум находится при пропускании 0,368 или оптической плотности [c.152]
Эти методы обстоятельно рассмотрены в работах [18, 19]. Изменяя способ измерения оптической плотности, часто можно понизить относительную ошибку определения концентрации, связанную с погрешностью прибора, до нескольких десятых нроцен- [c.153]
Для устранения случайных ошибок важно выбрать так величину регистрируемого сигнала, чтобы она значительно превышала уровень шумов. Поскольку при измерении пропускания (или оптической плотности) сравниваются два сигнала, важно, чтобы их разность тоже значительно превышала уровень шумов. Это приводит к определенным требованиям, которым должна удовлетворять величина пропускания исследуемого образца для того, чтобы относительная ошибка при ее определении была минимальной. Спек-тр9фотометры типа СФ-16 измеряют пропускание с точностью 1%. Относительная ошибка определения оптической плотности представлена на рис. 2.12, она имеет минимальное значение при значе-, ниях пропускания от 20 до 60%, что соот- [c.84]
Метод основан на измерении светопоглощения комплексов лантаноидов с кснленовым оранжевым при pH 5,6. Оптическую плотность измеряют на спектрофотометре при >. = 570 нм. Определению не мешают железо, алюминий, торий, титан, кальций и магний при их содержании до 2 мг. Относительная ошибка определения составляет 0,6%. [c.109]
Относительная ошибка определения концентрации этим методом уменьшается с увеличением концентрации С , раствора сравнения и получается наименьшей, когда светопоглощение или оптическая плотность исследуемого раствора и раствора сравнения одинаковы Сх = q). Практически концентрацию раствора сравнения выбирают так, чтобы значения оптической плотности, отвечающие разности концентраций исследуемого и нулевого растворов, лежали в оптимальной области измерений, т. е. от 0,3 до 0,7 Dom = 0,43) . Следует отметить, однако, что в фотоколориметрическом анализе увеличение концентрации С нулевого раствора не всегда приводит к повышению точности определения, главным образом, из-за возникающих отклонений от закона Бера (вследствие немонохроматич-ности поглощаемого света). Поэтому оптимальная концентрация нулевого раствора в каждом конкретном случае должна подбираться в зависимости от условий анализа и обеспечивать прохождение через окрашенный раствор достаточного количества света, для того чтобы можно было произвести установку гальванометра на пуль. Дифференциальный метод, в зависимости от способов измерения относительной оптической плотности исследуемого раствора и расчета его концентрации, может иметь несколько вариантов. [c.79]
В ряде случаев для повышения точности определений проводят фототурбидиметрическое титрование. Например, серу в органических соединениях после сожжения титруют раствором Ba lg в 90%-ном метаноле порциями по 0,5 мл. После добавления каждой порции раствор перемешивают и измеряют его оптическую плотность. Кривая зависимости оптической плотности от количества израсходованного Ba Ij состоит из двух линейных ветвей, точка пересечения которых соответствует точке эквивалентности. Относительная ошибка определения до 30 мкг S составляет + 10%, для количеств свыше 30 мкг S ошибка +2% [370]. [c.36]
Амиды Объемный и колориметрический методы количественного определения карбамида недостаточно чувствительны и требуют значительных затрат времени. Разработан спектрофотометрический метод, основанный на измерении оптической плотности окрашенного комплексного соединения, образующегося при реакции карбамида с п-диметиламино нзальдегидом Оптическая плотность измеротась на спектрофотометре СФ-4 при Х=420 нм в кюветах длиной 10 мм. Продолжительность опред ия 15 мин., относительная ошибка определения [c.44]
Постоянство нолярншс коэффициентов погашения для фосфора сохраняется в определенных пределах (относительная погрешность не цревышает 3,45 ) в интервале концентраций 0,15-1,00 мкг/мл, для кремния — в интервале концентраций 0,09-0,90 мкг/мл. При суммарной концентрации элементов выше 1,00 мкг/мл значения оптической плотности в максимуме поглощения цревншают 1. Это обстоятельство оцрелеляет верхний предел концентраций изучаемых элементов, так как относительная ошибка определения наименьшая, если значения лежат в интервале 0,3-1,0 [6]. [c.94]
Практикум по физической химии изд3 (1964) — [
c.101
]
Практикум по физической химии Изд 3 (1964) — [
c.101
]
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
Взамен ОФС ГФ X, ОФС ГФ XI, ОФС 42-0042-07 ГФ XII, ч.1
Уменьшение интенсивности монохроматического излучения, проходящего через гомогенную поглощающую среду, количественно описывается законом Бугера-Ламберта-Бера:
log10(1/Т) = А = ε ∙ c ∙ b ,(1)
где:
- Т – пропускание, отношение интенсивности светового потока, прошедшего через вещество, к интенсивности падающего на вещество светового потока; Т = I/I0;
- I – интенсивность прошедшего монохроматического излучения;
- I0 – интенсивность падающего монохроматического излучения;
- ε – молярный показатель поглощения;
- с – молярная концентрация вещества в растворе;
- b – длина оптического пути или толщина слоя, в сантиметрах.
Величина log10(1/Т) носит название оптической плотности, обозначается буквой А и является измеряемой величиной. В отсутствии других физико-химических факторов измеренная оптическая плотность (А) пропорциональна концентрации вещества в растворе (с) и толщине слоя (b).
Величина ![]() представляет собой удельный показатель поглощения, т.е. оптическую плотность раствора вещества с концентрацией 10 г/л (1 г/100 мл) в кювете с толщиной слоя 1 см. Величины
представляет собой удельный показатель поглощения, т.е. оптическую плотность раствора вещества с концентрацией 10 г/л (1 г/100 мл) в кювете с толщиной слоя 1 см. Величины ![]() и ε связаны соотношением:
и ε связаны соотношением:

где:
М.м. – молекулярная масса исследуемого вещества.
Измерение оптической плотности
Если нет других указаний в фармакопейной статье, измерение оптической плотности проводят при указанной длине волны с использованием кювет с толщиной слоя 1 см и при температуре (20 ± 1) °С по сравнению с тем же растворителем или той же смесью растворителей, в которой растворено вещество. При измерении оптической плотности раствора при данной длине волны оптическая плотность кюветы с растворителем, измеренная против воздуха при той же длине волны, не должна превышать 0,9 и, желательно, чтобы она была не менее 0,2.
Спектр поглощения представляют таким образом, чтобы оптическая плотность или ее некоторая функция были приведены по оси ординат, а длина волны или некоторая функция длины волны – по оси абсцисс.
Если в фармакопейной статье для максимума поглощения указывается только одна длина волны, то это означает, что полученное значение максимума не должно отличаться от указанного более чем на ± 2 нм.
Приборы
Спектрофотометры, предназначенные для измерений в ультрафиолетовой и видимой областях спектра, состоят из оптической системы, выделяющей монохроматическое излучение в области от 190 до 800 нм и обеспечивающей его прохождение через образец, и устройства для измерения оптической плотности.
Основными частями этих приборов являются: источник излучения, диспергирующий прибор (призма или решетка), щель для выделения полосы длин волн, кюветы для образцов, детектор излучаемой энергии, встроенные усилители и измерительные приборы.
Проверка шкалы длин волн в ультрафиолетовой и видимой области. Точность калибровки прибора по шкале длин волн в спектральном ряду проверяют по приведенным в табл. 1 спектральным линиям водородной (Hβ) или дейтериевой (Dβ) разрядной лампы, линиям паров ртути (Hg) кварцево-ртутной дуговой лампы, а также по максимумам поглощения раствора гольмия перхлората (Ho) (готовый реактив для калибровки спектрофотометра представляет собой 4 % раствор гольмия оксида в 14,1% растворе хлорной кислоты). Допустимое отклонение составляет ± 1 нм для ультрафиолетовой и ± 3 нм для видимой области.
Таблица 1 – Максимумы поглощения для проверки шкалы длин волн
| 241,15 нм (Но) | 404,66 нм (Hg) |
| 253,7 нм (Hg) | 435,83 нм (Hg) |
| 287,15 нм (Но) | 486,0 нм (Dβ) |
| 302,25 нм (Hg) | 486,1 нм (Нβ) |
| 313,16 нм (Hg) | 536,3 нм (Но) |
| 334,15 нм (Hg) | 546,07 нм (Hg) |
| 361,5 нм (Но) | 576,96 нм (Hg) |
| З65,48 нм (Hg) | 579,07 нм (Hg) |
Шкала длин волн может быть калибрована также при помощи подходящих стеклянных фильтров, которые имеют фиксированные полосы поглощения в видимой и ультрафиолетовой областях, а также стандартных стекол, содержащих дидим (смесь празеодима и неодима), и стекол, содержащих гольмий.
Проверка шкалы оптической плотности. Для проверки шкалы оптической плотности используют стандартные неорганические стеклянные фильтры или раствор калия дихромата при длинах волн, указанных в табл. 2, где для каждой длины волны приведено точное значение удельного показателя поглощения![]() и допустимые пределы.
и допустимые пределы.
Раствор калия дихромата для проверки шкалы оптической плотности при 235, 257, 313 и 350 нм готовят следующим образом: от 57,0 до 63,0 мг (точная навеска) калия дихромата, предварительно высушенного до постоянной массы при температуре 130 °С, растворяют в 0,005 М растворе серной кислоты и доводят объем раствора тем же растворителем до 1000 мл. Для проверки оптической плотности при 430 нм, растворяют 57,0-63,0 мг (точная навеска) калия дихромата в 0,005 М растворе серной кислоты и доводят объём раствора тем же растворителем до метки.
Таблица 2 ‑ Удельный показатель поглощения стандартов при различных длинах волн
| Длина волны, в нанометрах | Удельный показатель поглощения |
Допустимые пределы для |
| 235 | 124,5 | От 122,9 до 126,2 |
| 257 | 144,5 | От 142,8 до 146,2 |
| 313 | 48,6 | От 47,0 до 50,3 |
| 350 | 107,3 | От 105,6 до 109,0 |
| 430 | 15,9 | От 15,7 до 16,1 |
Предельный уровень рассеянного света. Рассеянный свет может быть обнаружен при данной длине волны с использованием соответствующих фильтров или растворов: например, оптическая плотность раствора 12 г/л калия хлорида в кювете с толщиной слоя 1 см резко увеличивается между 220 и 200 нм и должна быть больше 2 при 198 нм при использовании воды в качестве раствора сравнения.
Разрешающая способность (для качественного анализа). Если есть указание в фармакопейной статье, определяют разрешающую способность спектрофотометра следующим образом. Записывают спектр 0,02 % (об/об) раствора толуола в гексане. Минимально допустимое значение отношения оптической плотности в максимуме поглощения при 269 нм к оптической плотности в минимуме поглощения при 266 нм указывают в фармакопейной статье.
Ширина спектральной щели (для количественного анализа). В случае использования спектрофотометра с изменяемой шириной спектральной щели при выбранной длине волны возможны погрешности, связанные с шириной этой щели. Для их исключения ширина щели должна быть малой по сравнению с полушириной полосы поглощения (шириной на половине оптической плотности) и в то же время должна быть максимально велика для получения высокого значения интенсивности падающего монохроматического излучения (I0). Таким образом, ширина щели должна быть такой, чтобы дальнейшее ее уменьшение не изменяло величину измеряемой оптической плотности.
Кюветы. Допустимые отклонения в толщине слоя используемых кювет должны быть не более ±0,005 см. Кюветы, предназначенные для испытуемого раствора и раствора сравнения, должны иметь одинаковое пропускание (или оптическую плотность) при заполнении одним и тем же растворителем. В противном случае это различие следует учитывать.
Требования к растворителям. Для определений, производимых в ультрафиолетовой и видимой областях, образец анализируемого вещества растворяют в соответствующем растворителе, который должен быть оптически прозрачным в используемой области длин волн. Для этих областей длин волн пригодны многие растворители, в том числе вода, спирты, хлороформ, низшие углеводороды, эфиры и разбавленные растворы сильных кислот и щелочей.
Идентификация
Абсорбционную спектрофотометрию в ультрафиолетовой и видимой областях спектра применяют для определения подлинности лекарственных средств путем:
— сравнения спектров поглощения испытуемого раствора и раствора стандартного образца; в указанной области спектра должно наблюдаться совпадение положений максимумов, минимумов, плеч и точек перегиба;
— указания положений максимумов, минимумов, плеч и точек перегиба спектра поглощения испытуемого раствора; расхождение между наблюдаемыми и указанными длинами волн в максимумах и минимумах поглощения не должно обычно превышать ± 2 нм.
Возможны и другие варианты применения, оговоренные в фармакопейных статьях.
Количественное определение
Определение концентрации веществ спектрофотометрическим методом основано на использовании закона Бугера-Ламберта-Бера:

где:
С – концентрация вещества в г/100 мл;
А – оптическая плотность испытуемого раствора;
– удельный показатель поглощения вещества;
b – длина оптического пути или толщина слоя, в сантиметрах.
В ряде случаев, даже при использовании монохроматического излучения могут наблюдаться отклонения от закона Бугера-Ламберта-Бера, обусловленные процессами диссоциации, ассоциации и комплексообразования. Поэтому предварительно следует проверить линейность зависимости оптической плотности раствора от концентрации в аналитической области. При наличии отклонений от линейной зависимости следует пользоваться не формулой (3), а экспериментально найденной зависимостью.
Обычно определение концентрации спектрофотометрическим методом проводят с использованием стандартного образца. Расчет концентрации основан на использовании уравнения:

где:
С и С0 – концентрации испытуемого раствора и раствора стандартного образца, соответственно;
А и А0 – оптические плотности испытуемого раствора и раствора стандартного образца, соответственно.
Концентрации испытуемого и стандартного раствора должны быть близки.
Вначале измеряют оптическую плотность раствора стандартного образца, приготовленного, как указано в фармакопейной статье, затем проводят измерение оптической плотности испытуемого раствора. Второе измерение проводят сразу после первого, с использованием той же кюветы, в тех же экспериментальных условиях.
Метод с использованием стандартного образца является более точным и надежным. Возможность применения значения удельного показателя поглощения в каждом конкретном случае следует обосновывать. Обычно метод с использованием значения удельного показателя поглощения применим при допусках содержания анализируемого вещества не менее ±10 % от номинального содержания.
Многокомпонентный спектрофотометрический анализ
Многокомпонентный спектрофотометрический анализ (анализ смесей) применяют для одновременного количественного определения нескольких компонентов лекарственных средств, каждое из которых подчиняется закону Бугера-Ламберта-Бера.
Количественное определение в многокомпонентном спектрофотометрическом анализе основывается обычно на использовании уравнения:

где:
Аi – оптическая плотность испытуемого раствора при i-ой длине волны;
Еij – показатели поглощения (зависящие от способа выражения концентрации) j-го компонента образца при i-ой аналитической длине волны;
cj – концентрация j-го компонента образца.
Соответствующие методики проведения анализа и расчетные формулы указываются в фармакопейных статьях.
Производная спектрофотометрия
В производной спектрофотометрии исходные спектры поглощения (нулевого порядка) преобразуются в спектры производных первого, второго и более высокого порядков.
Спектр первой производной представляет собой график зависимости градиента кривой поглощения (скорость изменения оптической плотности от длины волны, dA/dλ) от длины волны.
Спектр второй производной представляет собой график зависимости кривизны спектра поглощения (d2A/dλ2) от длины волны. Вторая производная при любой длине волны связана с концентрацией следующим соотношением:

где:
А– оптическая плотность при длине волны λ;
![]() – удельный показатель поглощения при длине волны λ;
– удельный показатель поглощения при длине волны λ;
с– концентрация вещества в растворе, в граммах/100 мл;
l– толщина слоя, в сантиметрах.
Производная спектрофотометрия может быть использована как для целей идентификации веществ, так и для их количественного определения в многокомпонентных смесях, а также в тех случаях, когда имеется фоновое поглощение, вызванное присутствием веществ, содержание которых не регламентируется.
Приборы
Используют спектрофотометры, отвечающие указанным выше требованиям и оснащенные аналоговым резистивно-емкостным дифференцирующим модулем или цифровым дифференциатором, или другими средствами получения производных спектров, в соответствии с инструкцией к прибору. Некоторые методы получения спектров второй производной приводят к смещению длин волн относительно исходного спектра, что следует учитывать там, где это необходимо.
Разрешающая способность
Если указано в фармакопейных статьях, записывают спектр второй производной для раствора 0,2 г/л толуола в метаноле, используя метанол в качестве раствора сравнения. На спектре должен присутствовать небольшой отрицательный экстремум, расположенный между двумя большими отрицательными экстремумами при 261 нм и 268 нм, в соответствии с рис. 1. Если нет других указаний в фармакопейных статьях, отношение А/B должно быть не менее 0,2.
Методика
Процедура анализа аналогична применяемой в обычной спектрофотометрии, но вместо оптических плотностей используют производные. Готовят раствор испытуемого образца, настраивают прибор в соответствии с инструкцией производителя и рассчитывают количество определяемого вещества, как указано в фармакопейной статье.

Рисунок 1 – Спектр второй производной раствора толуола (0,2 г/л) в метаноле
Скачать в PDF ОФС.1.2.1.1.0003.15 Спектрофотометрия в УФ и видимой областях